

Summer Diet of South China Sika Deer (Cervus nippon kopschi) in Taohongling based on High-Throughput Sequencing and Metabarcoding trnL





Summer Diet of South China Sika Deer (Cervus nippon kopschi) in Taohongling based on High-Throughput Sequencing
and Metabarcoding trnL
Yongtao Xu1, Dandan Wang1, Xiaolong Hu1, Minling Li1, Ming Tang1,
Wuhua Liu2, Jianwen Zhan2 and Weiwei Zhang1*
1College of Forestry, Jiangxi Agricultural University, Nanchan, 330045, China.
2Taohongling National Nature Reserve of Sika Deer, Pengze 332700, China.
Yongtao Xu and Dandan Wang contributed equally to this study.
ABSTRACT
The sika deer (Cervus nippon) is listed as a Class I National Key protected wild animal and is an endemic species in the East Asian monsoon region. Although the species as a whole is thriving, it is endangered and locally extinct in many areas of China, and the South China sika deer (Cervus nippon kopschi) is one of three subspecies left in China. Diet analysis is one of the core contents in studying animal habitat requirements, and a study of their diet could provide valuable reference for species conservation and management. The metabarcoding trnL regions of the chloroplast genome from 30 fecal samples of Taohongling sika deer were amplified with universal primers and sequenced by high-throughput. We found that 28 out of 30 valid samples had a total of 677,856 valid amplified sequences with an average valid sequence of 24,209.14±323.83. A total of 326 OTUs were obtained from 28 valid samples, and the MP, SS, and NJS groups shared 222 OTUs, accounting for 68.09% of the total OTUs. OTUs alignment indicated that the forage plants of sika deer belong to 82 families, and 110 genera. An alpha diversity analysis showed that there was no significant difference (p > 0.05). The NMDS analysis found considerable overlap at the three sampling sites, and the niche breadths of SS (Fir forests), MP (The nursery base), and NJS (Nie Jiashan) were 9.593, 9.426, and 9.419, respectively. High-throughput sequencing and metabarcoding trnL could provide higher taxonomic resolution, identify more food items, and can simultaneously analyze a larger number of samples compared with traditional diet methods. This study assembled basic summer diet data and provided an important accumulation for the protection and monitoring of Taohongling sika deers, which also provides reference for the establishment of local database in the future.
Article Information
Received 29 June 2022
Revised 15 September 2022
Accepted 08 October 2022
Available online 05 January 2023
(early access)
Published 29 January 2024
Authors’ Contribution
YX and DW performed the experiments, analyzed the data, prepared figures and/or tables, and approved the final draft. XH, ML, MT, WL and JZ analyzed the data. WZ conceived and designed the experiments, analyzed the data, prepared figures and/or tables and approved the final draft.
Key words
Cervus nippon kopschi, Diet analysis, Metabarcoding, OTUs, Summer
DOI: https://dx.doi.org/10.17582/journal.pjz/20220629000640
* Corresponding author: [email protected]
0030-9923/2024/0002-0771 $ 9.00/0
Copyright 2024 by the authors. Licensee Zoological Society of Pakistan.
This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Introduction
The sika deer (Cervus nippon) or spotted deer, belongs to the family Cervidae. It is a ruminant endemic species in the East Asian monsoon region (Guo, 2000). Sika deer was listed as a first class national protected animal in 1989 (Fu et al., 2006), and EN (Endangered) species on China’s red list of biodiversity in 1998 (Wang, 1998). They inhabit temperate and subtropical woodlands, often in areas suitable for farming and other human exploitation. Due to indiscriminate poaching and a reduction in natural habitat over a long period, the wild sika deer population has dropped sharply (Guo, 2001). Historically, there were 6 subspecies of wild sika deer in China; Cervus nippon grassianus of Shanxi, Ningxia and eastern Gansu provinces, Cervus nippon mandarinus of Hebei, Shandong and northern Henan provinces, and Cervus nippon taiouanus of Taiwan province died out in the 1940s (He et al., 2019), and three other remained subspecies including Cervus nippon hortulorum of Heilongjiang and Jilin provinces, Cervus nippon sichuanicus of Sichuan province, and Cervus nippon kopschi were listed EN species. The South China sika deer (Cervus nippon kopschi) is mainly distributed in Taohongling of Jiangxi Province, which represents the largest population of 400 individuals compared with isolated populations in Tianmushan, Huanggunshan, Dahuishan, western Jingxian and Qianxian (Gao et al., 2009).
Food provides necessary energy and nutrients for the survival of a species. Diet research is the basis of animal nutrition and the premise of population ecology research. At present, the common research methods of researching herbivore feeding habits mainly include indoor cage feeding, gastric content analysis, fecal analysis, and direct observation of utilization. Most of these traditional dietary analysis methods are based on morphological identification (Kaneko and Lawler, 2006), near-infrared spectral reflectance (Holechek et al., 1982; Heroldova et al., 2010) or isotopic principles (Wang et al., 2015; Zheng et al., 2015). Many ecological problems related to feeding habits have been solved, but there are still some deficiencies: the results are largely qualitative descriptions, some animals are difficult to track (Shao et al., 2019), ingestion is incomplete or related to specific microstructures, and the identification of foods with similar morphology is challenging (Lu et al., 2019). The accuracy and precision of the food habit analysis method directly impacts the exploration of related theories of food habits and the practice of applying the results of food habits to wildlife protection. With the development of high-throughput sequencing, this technology has been gradually extended to the analysis of food habits of wild animals (Sun et al., 2021), which has greatly improved the efficiency of food analysis and broadened the scope of food analysis.
High-throughput sequencing technologies have transformed our ability to explore complex environmental samples. Due to the characteristics of high sensitivity and the ability to process a large amount of data, high-throughput sequencing technology can generate millions of reads on a sequencing platform (Liu et al., 2018), and its high efficiency is preferred by researchers. Due to the partial degradation of food samples (in feces, boluses or gastric contents), and because the PCR amplification efficiency of short fragments is higher than that of long fragments, the read length range of high-throughput sequencing is very suitable for food analysis and comparison with databases. After pairing, the classification of foods can be refined to species-level taxa, and the relative amount and preference of animals for a food can also be judged based on the number of DNA sequences. The food selection of herbivores is the most active field of foraging ecology research (Li and Liu, 2003). In practice, dietary research is of great significance for the protection and management of species, particularly endangered and rare species, as well as for domestication and reproduction (Guo et al., 2021).
Over the years, Taohongling National Nature Reserve has implemented strict closure of the mountains for afforestation. The natural succession of vegetation has annually reduced the area of habitat suitable for sika deer, and the vegetation in many areas has been restored to evergreen and deciduous broad-leaved mixed forests (Wang, 2018). However, the most suitable habitat for wild sika deer contains shrubs and grasses (Gao et al., 2009). Taohongling Reserve is facing the problem of overly dense shrubs and arborization, which seriously affects the habitat and population growth of sika deer. To effectively improve the habitat of the nature reserve, formulate a reasonable vegetation plan, delineate key foraging protection areas and establish animal activity corridors, there is an urgent need to understand the quantity and types of existing food resources of wild sika deers. Diet reflects the use of food resources by animals, and acquisition of food composition accurately is the premise of fully understanding the living habits and food web relationships of this species (Liu et al., 2018; Lu et al., 2019). Thus this study is of great significance to the assessment of forage resources for sika deers as consumers in the ecosystem.
Materials and methods
Sample collection
The Taohongling Sika Deer National Nature Reserve (29°42′-29°53′N, 116°32′-116°43′E) is the main distribution area of sika deer subspecies in South China (Wu et al., 2012), and is mainly located along the middle and lower reaches of the Yangtze River in Pengze County, Jiangxi Province. The total area is 125 km2, the core area is 26.7 km2, and the terrain is a low mountain and hilly area with an elevation of 100-500 m (Zhou et al., 2019). From July to August 2020, we collected 30 fecal samples of South China sika deer from nursery bases (hereafter MP), fir forests (hereafter SS), and NieJiashan (hereafter NJS) occurred frequently by infrared cameras in the Jiangxi Taohongling Sika Deer National Reserve. We brought them back to the laboratory for storage at -80 °C. Based on the morphological identification, the collected samples similar to black peanuts can represent this species (Fig. 1).
Genomic DNA extraction and trnL amplification
We selected and mixed five fecal pellets as one final sample using Liquid nitrogen grounding method (Yang, 2016). The total DNA of sika deer feces was extracted using the plant genome extraction kit, and the A260/A280 of most DNA extracts was between 1.70 and 2.28, indicating that the extraction was effective. In this study, the plant chloroplast trnL intron metabarcoding universal primer sequences c: CGAAATCGGTAGACGCTACG and h: CCATTGAGTCTCTGCACCTATC were used to amplify DNA by approximately 145 bp (Parameswaran et al., 2007). This primer pair was chosen because it can amplify DNA from a wide range of plants. PCR amplifications were carried out in a total volume of 25 μl containing 12.5 μl PCR mix (Tiangen, Beijing, China), 1 μl DNA, 1 μl of each primer, and 9.5 μl H2O. The reaction conditions were as follows: Denaturation at 95℃ for 3 min followed by 35 cycles at 95℃ for 30 s, 56℃ for 30 s, and 72℃ for 45 s with a final 10 min at 72℃ and storage at 4℃ for 10 h (Yang, 2016). Agarose gel electrophoresis found that the bands of samples 6 and 28 were correct, but the low concentration could not be used in subsequent experiments (Hou et al., 2021).
Sequencing and data processing
The PCR products of 28 fecal DNA samples were purified and mixed repeatedly and sequenced by Shenzhen Microsun Technology Co., Ltd. Paired-end sequencing was used to complete the sequencing based on the Illumina HiSeq X Ten system (Illumina Inc., San Diego, CA, USA), and each end read was 150 bp in length. FLASH (V0.33, http://www.usadellab.org/cms/?page=trimmomatic) and Trimmomatic software (V1.2.11, https://ccb.jhu.edu/software/FLASH/) were used for raw sample statistics and effective data statistics. The data of each sample were distinguished based on the index sequence, and the extracted data were saved in FASTQ format. Each sample of PE data had two files, fq1 and fq2, which are the reads at both ends of the sequencing, and the sequences correspond one-to-one in order. MiSeq sequencing generates paired-end sequence data. First, based on the overlap between the PE reads, the paired reads were spliced (merged) into a sequence, and the quality of the reads and the effect of the merge were filtered for quality control. The barcode at the end and the primer sequence distinguished the sample to obtain an effective sequence and correct the sequence direction, which is the optimized data. The data decontamination method and parameters were as follows. Bases with a tail quality value below 20 were filtered, and a 50 bp window was set. If the average quality value in the window was below 20, the back-end bases were truncated from the window, and the bases below 50 bp were filtered after quality control. Reads containing N bases were removed; based on the overlap between PE reads, the paired reads were spliced (merged) into a sequence, the minimum overlap length was 10 bp, and the maximum mismatch ratio allowed in the overlap region of the splicedsequence was 0.2. These were screened for nonconforming sequences; the samples were distinguished based on the barcodes and primers at the beginning and end of the sequence, and the sequence direction was adjusted. The number of mismatches allowed by barcodes was 0, and the maximum number of primer mismatches was 2 (Magoc and Salzberg, 2011; Bolger et al., 2014).
Statistical analysis
The original data obtained by sequencing contained a certain proportion of interference data. To ensure the accuracy of the analysis, the original data were first spliced and filtered to obtain valid data. Based on the effective data, the sequences with a similarity of 97% were combined, the OTUs (Operational Taxonomic Units) were divided, and the sequence with the highest abundance in each OTUs was selected as the representative sequence of the OTUs; these sequences were compared with the NCBI database, and the plant taxonomic status of the representative sequence of the OTUs was identified by GenBank. The alpha diversity index of the sample was calculated based on the sequence number to obtain information on species richness and evenness within the sample, common and unique OTUs information among different samples or groups, etc. The Kruskal–Wallis test was used to assess the difference in alpha diversity between different sample groups, and the relationship between samples was shown by drawing a heat map. The analysis of alpha and beta diversity was mainly performed with the QIIME2 diversity plugin. Alpha diversity is an indicator that reflects the number of species and the relative abundance of species in a community or habitat. The alpha diversity Sparse curve (rarefaction curve) is constructed based on the amount of sequencing data and species diversity, and used to illustrate whether the amount of sequencing data in the sample is adequate and indirectly reflects the abundance of species in the sample. The alpha diversity index includes two factors: The richness and evenness of species composition in the samples. The chao1, faith_pd, observed_OTUs, Shannon index, and Simpson were used to reflect the richness, and the Pielou evenness index is maximized when all species have the same number of individuals and is minimized when all individuals belong to one species. Food source plant diversity analysis was conducted as follows.
The Shannon–Wiener diversity index (H’) was calculated to explore the diet diversity of each sample site (Smith, 1982). The following equations were used:
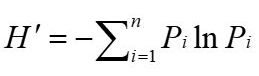 ....(1)
....(1)
Pielou evenness index (Pielou, 1969):
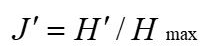 ....(2)
....(2)
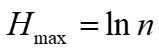 ....(3)
....(3)
Where n is the number of plant species in the fecal sample, and the number of plant species is represented by the number of plant OTUs types present.
The dietary breadth (B) was measured using Levins’ index (Levins, 1968), according to the following formula:
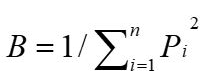 ...(4)
...(4)
Where Pi is the proportion of plant item i out of all foods.
Beta diversity refers to the dissimilarity of species composition between different communities along an environmental gradient or the replacement rate of species along an environmental gradient, also known as between-habitat diversity. Bray-Curtis distance was performed to investigate the structural variation of forage plant communities across sampling sites and then visualized via nonmetric multidimensional scaling (NMDS) (Vázquez-Baeza et al., 2013). The Bray-Curtis distance was calculated based on the OTUs abundance. A smaller value indicates a smaller difference in species diversity between the two samples. Nonmetric multidimensional scaling (NMDS) is an indirect gradient analysis method which produces an ordination based on a distance or dissimilarity matrix.
Results
TrnL metabarcoding sequencing and OTUs statistics
A total of 677,856 original amplified reads were obtained from the 28 valid samples using trnL metabarcoding after high throughput sequencing, two samples were eliminated that did not meet the sequencing requirements. The total number of valid bases was 98,785,525, the shortest average reads were 120.46±0.2 bp, the longest average reads were 209.89±10.80 bp, and the total average was 145.65±1.22 bp (Table I). OTUs annotation of all valid sequences from the 28 valid samples were clustered/ denoised to form OTUs, which can also be called characteristic sequences. The representative sequences of OTUs were selected and compared with the NT database based on 99% sequence similarity clustering to obtain species annotation information.
Table I. trnL amplicon sequencing information of South China Sika deer based on high-throughput.
|
Sampling sites/ sample ID |
Raw_reads |
Bases |
Avg (bp) |
Min (bp) |
Max (bp) |
|
SS |
|||||
|
SS01 |
24,433 |
3,618,859 |
148.11 |
120 |
331 |
|
SS02 |
20,895 |
2,755,403 |
131.86 |
120 |
172 |
|
SS03 |
23,998 |
3,242611 |
135.12 |
121 |
175 |
|
SS04 |
23,359 |
3,507,417 |
150.15 |
120 |
212 |
|
SS05 |
24,780 |
3,477,121 |
140.31 |
120 |
189 |
|
SS06 |
24,303 |
3,420,633 |
140.74 |
120 |
170 |
|
SS07 |
23,125 |
3,453,586 |
149.34 |
120 |
334 |
|
SS08 |
24,911 |
3,247,497 |
130.36 |
120 |
318 |
|
SS09 |
24,436 |
3,196,823 |
130.82 |
120 |
177 |
|
MP |
|||||
|
MP01 |
23,127 |
3,452,138 |
149.26 |
120 |
172 |
|
MP02 |
23,707 |
3,533,446 |
149.04 |
120 |
187 |
|
MP03 |
24,811 |
3,704,635 |
149.31 |
123 |
180 |
|
MP04 |
23,648 |
3,501,489 |
148.06 |
120 |
188 |
|
MP05 |
24,034 |
3,568,288 |
148.46 |
120 |
180 |
|
MP06 |
31,921 |
4,801,496 |
150.41 |
120 |
224 |
|
MP07 |
24,593 |
3,685,284 |
149.85 |
123 |
180 |
|
MP08 |
24,982 |
3,723,175 |
149.03 |
120 |
198 |
|
MP09 |
23,115 |
3,445,765 |
149.07 |
123 |
188 |
|
MP10 |
23,468 |
3,577,097 |
152.42 |
120 |
185 |
|
NJS |
|||||
|
NJS01 |
23,873 |
3,446,368 |
144.36 |
120 |
177 |
|
NJS02 |
23,892 |
3,575,334 |
149.64 |
120 |
187 |
|
NJS03 |
24,954 |
3,742,729 |
149.98 |
123 |
180 |
|
NJS04 |
23,928 |
3,536,992 |
147.81 |
120 |
187 |
|
NJS05 |
23,884 |
3,579,626 |
149.87 |
120 |
178 |
|
NJS06 |
23,982 |
3,524,254 |
146.95 |
120 |
187 |
|
NJS07 |
24,023 |
3,612,081 |
150.35 |
120 |
329 |
|
NJS08 |
23,911 |
3,352,202 |
140.19 |
120 |
166 |
|
NJS09 |
23,763 |
3,503,176 |
147.42 |
120 |
326 |
|
- |
677,856 |
98,785,525 |
4,078.29 |
3,373 |
5,877 |
|
- |
24,209.14 ±323.83 |
3528054.46 ±6003215 |
145.65± 1.22 |
120.46 ±0.20 |
209.89 ±10.80 |
The special and common OTUs between samples were identified to reflect the compositional similarity of samples at the OTUs level, and the differences and similarities in the community composition of forage plants in the different habitats of sika deers. Petal map analysis can illustrate the number of OTUs that are unique or shared between different sample groups; the center shows the number of OTUs shared by all groups, and the remaining areas show the number of OTUs unique to each individual group. The petal map showed that the MP, SS and NJS sampling sites shared 222 OTUs, accounting for approximately 68.09% of the total OTUs (326). Among the sites, the MP group had 26 unique OTUs, accounting for approximately 7.97% of the total OTUs; the SS group had 57 unique OTUs, accounting for approximately 17.48% of the total OTUs; and the NJS group had 21 unique OTUs, accounting for approximately 6.44% of the total OTUs (Fig. 2).
In the OTUs sequences obtained in this study, there were point mutations among multiple OTUs representative sequences belonging to the same species. Among the OTUs sequences obtained in this study, BLAST alignment showed that some of the different OTUs were due to intraspecific genetic variation. For example, by Blast comparison, OTUs 451, OTUs 30, OTUs 277, OTUs 270, OTUs 255, OTUs 253, OTUs 247, OTUs 246, OTUs 221, OTUs 213, OTUs 206, OTUs 170 and OTUs 104 all represent Smilax. Taking Smilax as an example, MEGA alignment and DnaSP analysis showed that except for OTUs 234, which had 2 base deletions of 148 bp, the other OTUs were 150 bp in length, the number of polymorphic sites was 25, the haplotype diversity variation index was 0.00150, and the nucleotide diversity index (Pi) was 0.03907. Among the types of base substitution, base conversion was the main type accounting for 47.82% of substitutions, base trans version accounted for 43.47% of substitutions, and the combined types accounted for 8.71% of substitutions. In summary, the number of species identified was less than the number of OTUs.
Diet diversity and niche breadth in summer
The OTUs rarefaction curves of all samples tend to be flat, and the readings are all above 20,000, indicating that the amount of sequencing was reasonable, and the sequencing depth was sufficient (Fig. 3). Alpha diversity analysis of different samples groups showed that NJS group had higher chao 1 (137.60±21.95), faith_pd (7.72± 1.12), observed_OTUs (112.56±16.81), shannon (3.01±0.43), simpson (0.70±0.08) and pileou indices (0.62±0.09) than those of MP and SS groups (Table II). Moreover, no statistically significant difference among the different sampling groups was found using Wilcoxon test (p > 0.05) (Table III).
Patterns of diet composition of three sampling sites were performed by NMDS analysis based on the Bray-Curtis dissimilarity. The dots in the figure represent samples, the dots with different colors belong to different samples (groups), and the distance between the dots indicates the degree of difference between the samples. A shorter distance between two dots indicates that the two samples have a similar community structure. A higher degree of difference indicates less similarity, the results showed that the community structure of MP and NJS samples was more similar in Figure 4.
Table II. Alpha diversity index including chao1, faith_pd, observed_otus, shannon, simpson, and pileou in three sampling groups.
|
Sample ID |
Chao 1 |
Faith_pd |
Observed otus |
Shannon |
Simpson |
Pileou |
|
SS01 |
147.15 |
5.25 |
134 |
3.39 |
0.80 |
0.67 |
|
SS02 |
68.27 |
6.51 |
60 |
2.02 |
0.57 |
0.40 |
|
SS03 |
176.3 |
5.89 |
143 |
2.92 |
0.65 |
0.58 |
|
SS04 |
196.06 |
12.38 |
163 |
4.50 |
0.92 |
0.89 |
|
SS05 |
38.87 |
2.78 |
37 |
1.27 |
0.35 |
0.25 |
|
SS06 |
177.83 |
7.75 |
152 |
3.99 |
0.87 |
0.79 |
|
SS07 |
174.23 |
10.99 |
152 |
3.87 |
0.84 |
0.77 |
|
SS08 |
48.5 |
5.84 |
44 |
1.93 |
0.51 |
0.38 |
|
SS09 |
103 |
6.54 |
69 |
1.70 |
0.49 |
0.33 |
|
Mean ±SE |
125.58 ± 20.53 |
7.10 ± 0.98 |
106 ± 17.37 |
2.84 ± 0.39 |
0.67 ± 0.06 |
0.56 ± 0.077 |
|
MP01 |
134.6 |
7.81 |
127 |
2.87 |
0.66 |
0.58 |
|
MP02 |
197.35 |
10.81 |
156 |
4.20 |
0.90 |
0.86 |
|
MP03 |
58 |
3.17 |
49 |
2.11 |
0.67 |
0.43 |
|
MP04 |
107.87 |
7.33 |
79 |
3.40 |
0.84 |
0.69 |
|
MP05 |
93.5 |
4.11 |
68 |
2.08 |
0.56 |
0.42 |
|
MP06 |
51 |
3.01 |
44 |
1.20 |
0.33 |
0.24 |
|
MP07 |
45 |
2.23 |
39 |
1.44 |
0.40 |
0.29 |
|
MP08 |
176.07 |
9.96 |
136 |
4.05 |
0.89 |
0.83 |
|
MP09 |
198.25 |
6.14 |
147 |
4.12 |
0.88 |
0.84 |
|
MP10 |
40.11 |
5.16 |
37 |
1.10 |
0.37 |
0.22 |
|
Mean ± SE |
110.18 ± 19.99 |
5.973 ± 0.94 |
88.2 ± 15.23 |
2.66 ± 0.39 |
0.65 ± 0.07 |
0.54 ± 0.08 |
|
NJS01 |
91.11 |
6.74 |
70 |
2.68 |
0.74 |
0.55 |
|
NJS02 |
191.25 |
11.70 |
168 |
4.28 |
0.90 |
0.88 |
|
NJS03 |
52 |
3.15 |
45 |
1.16 |
0.30 |
0.23 |
|
NJS04 |
188.61 |
10.49 |
148 |
4.07 |
0.89 |
0.83 |
|
NJS05 |
52 |
3.53 |
45 |
1.06 |
0.26 |
0.21 |
|
NJS06 |
190.2 |
11.84 |
165 |
4.40 |
0.92 |
0.90 |
|
NJS07 |
130.27 |
9.75 |
113 |
3.20 |
0.78 |
0.65 |
|
NJS08 |
111.17 |
5.41 |
100 |
2.34 |
0.68 |
0.48 |
|
NJS09 |
231.76 |
6.90 |
159 |
3.94 |
0.86 |
0.81 |
|
Mean ± SE |
137.60 ± 21.95 |
7.72 ± 1.12 |
112.56 ± 16.81 |
3.01 ± 0.43 |
0.70 ± 0.08 |
0.62 ± 0.09 |
Table III. Multiple comparisons of Alpha diversity index using wilcoxon method.
|
Variable |
Group 1 |
Group 2 |
P. format |
P. signif |
Method |
|
chao1 |
SS |
MP |
0.66 |
ns |
Wilcoxon |
|
chao1 |
SS |
NJS |
0.93 |
ns |
Wilcoxon |
|
chao1 |
MP |
NJS |
0.45 |
ns |
Wilcoxon |
|
faith_pd |
SS |
MP |
0.4 |
ns |
Wilcoxon |
|
faith_pd |
SS |
NJS |
0.86 |
ns |
Wilcoxon |
|
faith_pd |
MP |
NJS |
0.28 |
ns |
Wilcoxon |
|
observed_otus |
SS |
MP |
0.37 |
ns |
Wilcoxon |
|
observed_otus |
SS |
NJS |
0.82 |
ns |
Wilcoxon |
|
observed_otus |
MP |
NJS |
0.24 |
ns |
Wilcoxon |
|
shannon |
SS |
MP |
0.9 |
ns |
Wilcoxon |
|
shannon |
SS |
NJS |
0.8 |
ns |
Wilcoxon |
|
shannon |
MP |
NJS |
0.6 |
ns |
Wilcoxon |
|
simpson |
SS |
MP |
0.97 |
ns |
Wilcoxon |
|
simpson |
SS |
NJS |
0.67 |
ns |
Wilcoxon |
|
simpson |
MP |
NJS |
0.6 |
ns |
Wilcoxon |
|
Pileou |
SS |
MP |
0.93 |
ns |
Wilcoxon |
|
Pileou |
SS |
NJS |
0.66 |
ns |
Wilcoxon |
|
Pileou |
MP |
NJS |
0.62 |
ns |
Wilcoxon |
Overall, 99.96% of the sequences from sika deer feces were identified at the family level, and 99.03% were identified at the genus level. In 28 samples of sika deer feces, a total of 326 OTUs were obtained, representing plants from 82 families, 110 genera. The 18 most common families in the sika deer diet were Smilacaceae, Rosaceae, Poaceae, Asteraceae, Hamamelidaceae, Polygonaceae, Bryaceae, Ericaceae, Anacardiaceae, Dicranaceae, Fabaceae, Oxalidaceae, Fagaceae, Phyllanthaceae, Caprifoliaceae, Staphyleaceae, Zygnemataceae, and Rhabdoweisiaceae. Moreover, all the information of family, genus and the OTUs number in diet analysis were upload as Supplementary Materials. The 18 most common genera in the sika deer diet were Smilax, Rosa, Loropetalum, Persicaria, Rosulabryum, Rhododendron, Rhus, Solidago, Dicranella, Setaria, Digitaria, Bidens, Drepanostachyum, Oxalis, Rubus, Oxychloris, Quercus, Wisteria and two unidentified items with higher abundance were showed in Figure 5. The dietary niche reflected the ability of species to utilize resources, and we found the niche breadths of SS, MP and NJS were 9.593, 9.426, and 9.419 using Levins’ index, respectively. These results showed that there was no significant difference in niche breadth among the three regions (p > 0.05).
Discussion
TrnL outperforms rbcL as a DNA metabarcoding marker
DNA barcoding is a relatively new concept, aiming to provide rapid, accurate and automatable species identifications by using a standardized DNA gene region as a tag. Eight candidate genes are commonly used in the diet identification of herbivores: rbcL, rpoC1, rpoB, matK, trnH-psbA, trnL, atpF-atpH, and psbK-psbI. In terms of single-gene barcoding, we should choose the most suitable DNA plant barcoding markers. Four out of eight candidate genes were screened and indicated that trnL amplified more successfully than rbcL for waterbirds in Shengjin Lake during the winter with success rates of 100% and 91%, respectively (Yang, 2016). Diet analysis of white-faced capuchin monkeys in Costa Rica also supported that trnL was more reliable than rbcL for plant analysis and could be used to quantitatively assess intra- or interspecific diet differences (Mallott et al., 2018).
In this study, we also chose trnL for the diet analysis of sika deers. Using the chloroplast trnL (UAA) intron (254-767bp), the primers were found to be highly conserved, and the amplification system was very robust. The P6 loop (10-143bp) can even be amplified when using highly degraded DNA from processed food or from permafrost samples and has the potential to be extensively used in the food industry, in forensic science, in diet analysis based on feces and in ancient DNA studies. Although the entire trnL intron for DNA barcoding has limited resolution, it is much higher in specific contexts such as with species originating from a single ecosystem or commonly eaten plants. Appropriate barcode gene fragments should be selected based on the characteristics of the research object. Typically, mitochondria are chose for carnivores, chloroplasts are chosen for herbivores, and omnivores need to consider a synthesis of approaches (Tarberlet et al., 2007; Liu et al., 2018; Tercel et al., 2021).
Food composition, preferences, and niche breadth of sika deer
Detailed knowledge of diet composition and preferred food items is important for exploring optimal foraging theory, which assumes that the main goal of a generalist herbivore is to maximize the quantity or quality of food while foraging or to obtain food efficiently while avoiding predation (Goldberg et al., 2020). Compared with tracking observation in Sichuan sika deer (Guo, 2001) and artificial feeding method in captive sika deer in Northeast China (Zhang et al., 2011), more diet varieties were found in Taohongling sika deers, which may be due to the rapid growth rate of plants in the summer when buds are tender and nutritious; it may also be related to the high-throughput sequencing method, which can reflect the feeding situation more comprehensively. Existing studies have shown that 12 independent fecal samples could record 95% of the diet information (Mata et al., 2019), and thus the screening results of 28 samples in this study could reflect the diet of sika deers appropriately in the summer.
Wildlife foods can be categorized as bulk foods, favorite foods, and emergency foods. Those with a higher cumulative abundance of OTUs can be regarded as a favorite food or a bulk food, and those with a lower abundance can be regarded as occasional or emergency food. Taking Smilax as an example, it can be speculated that this is a favorite plant of sika deer. Smilax contains saponins and alkaloids, which are sweet and warm and have the properties of expelling wind and promoting blood circulation. The roots, leaves, flowers and fruits of Rhus can be used for medicinal purposes. The stalks and leaves of Digitara can be used as food and can also be used as medicine to treat carbuncle stasis and ringworm. A commonly used herbal medicine in China is Bidens, which has the properties of clearing away heat, detoxification, dissipating blood stasis and promoting blood circulation. We also identified algal plants, i.e., Vaucheria and Botrydiopsis, which may have been obtained from drinking water. The summer dietary niche breadth of sika deer is higher than autumn diet of alashan red deer (Xu et al., 2018), spring diet of helanshan red deer (Wang et al., 2019) and qionglai mountains sambar (Zhang et al., 2020). It is speculated that the abundant plant resources and diverse habitat types in Taohongling nature reserve may facilitate the flexible foraging strategies for sika deers.
Unidentified plant species
In the OTUs obtained in this study, point mutations occurred within species among multiple representative OTUs. Accumulation and merging were performed when calculating the abundance ratio; therefore, the number of OTUs was greater than the number of plant species. However, not all the OTUs could be aligned to a specific plant species in the NCBI database. In this study, the lack of ferns in the diets of sika deers reflected that trnL could not be used for the identification of ferns due to mismatch (Nakahama et al., 2021). The rbcL and matK regions of chloroplast DNA can be used for the identification of ferns (Li et al., 2011), and the subsequent combination of molecular markers will help to understand the feeding habits of sika deer in more detail.
We speculate that due to the existence of variability, limitations on amplification efficiency and gene deletion, it is difficult to identify all plant species using only a single gene fragment, so it is necessary to add auxiliary barcodes, i.e., to use DNA metabarcoing multiplexing. The following combined barcodes of chloroplast genomic regions have been reported: rbcL + trnH-psbA, rbcL + ITS2, rpoC1 + matK + trnH-psbA, and rpoC1 + matK + rpoB. For plant species with higher abundance that cannot be determined or identified, the integration of multimarker met barcoding may help improve accuracy. The recognition rate of these combinations is higher than that of a single gene barcode, however, using multiple markers need to overcome the problems of primer specificity, bias, mutual interferences, and sequencing cost (Da Silva et al., 2019; De Barba et al., 2014). In addition, if new plant species are not recorded or the database is not available, it may be necessary to improve the public database or build a local database for comparison. In our study, the unique OTUs of the three sampling sites can evaluate the importance of species and the distribution of potential forage plants in the sampling habitat and provide investigation and monitoring directions for the documentation of plant resources in the nature reserve.
At present, the application of high-throughput technology for diet analysis is just emerging, and the diets of most animals are still in the relatively rough qualitative description stage. As long as researchers can reasonably control the sources of error of high-throughput sequencing and select appropriate DNA metabarcoding for amplification (De et al., 2014; Brown et al., 2014), this technology will have advantages over other dietary analysis methods. With this technology, we can explore the relationship between animals feeding on plants and the ecological processes of plant pollination and seed dispersal, determine the relationship between the pollination process and its mediation by vector animals, place animals in their ecological role within the network structure (Valentini et al., 2009), study the impact of climate change on animal dietary preferences and shifts, predict animal preferences for foods that are affected by climate change, and simulate how animals will adapt to global warming by shifting their diets (Carreira et al., 2016). In summary, it can open new perspectives in ecology, not only by allowing large scale studies on diet, but also enhancing studies on resource partitioning among competing species, and describing food webs in ecosystem.
Conclusion
Diet research is of great significance for the protection, management, domestication and reproduction of rare and endangered species. Our study assembled basic data on Taohongling sika deer in the summer using high-throughput sequencing and metabarcoding technology, and provided an important reference for the diet of sika deer in protecting and monitoring, which would facillitate the habitat management to improve the availability of foraging resources.
Acknowledgment
This research was supported by the Science and Technology Program of Jiangxi Provincial Department of Education (GJJ180225) and the National Natural Science Foundation of China (31960118). We are grateful to Xiaohong Liu, Yongjiang Chen and Yulu Chen of Taohongling Sika Deer National Nature Reserve for their help in sample collection, and Dr Hao Zang for the help with R analysis. The authors also thank Microeco Tech Co., Ltd. Shenzhen China for sequencing service and assistance.
Funding
Financial support was provided by the Science and Technology Program of Jiangxi Provincial Department of Education (GJJ180225) and the National Natural Science Foundation of China (31960118).
Ethical statement
No animals were captured, and fecal samples analysis were performed based on the noninvasive principle. We conducted the research under the guidelines of Taohongling Sika Deer National Nature Reserve and Wildlife Conservation Research Center, Jiangxi Agricultural University.
There is supplementary material associated with this article. Access the material online at: https://dx.doi.org/10.17582/journal.pjz/20220629000640
Statement of conflict of interest
The authors have declared no conflict of interest.
References
Bolger, A.M., Lohse, M., and Usadel, B., 2014. Trimmomatic: A flexible trimmer for Illumina Sequence Data. Bioinformatics, 30: 2114-2120. https://doi.org/10.1093/bioinformatics/btu170
Brown, D.S., Ebenezer, K.L., and Symondson, W.O.C., 2014. Molecular analysis of the diets of snakes: Changes in prey exploitation during development of the rare smooth snake, Coronella austriaca. Mol. Ecol., 23: 3734-3743. https://doi.org/10.1111/mec.12475
Carreira, B.M., Segurado, P., Orizaola, G., Gonçalves, N., Pinto, V., Laurila, A., and Rebelo, R., 2016. Warm vegetarians? Heat waves and diet shifts in tadpoles. Ecology, 97: 2964-2974. https://doi.org/10.1002/ecy.1541
Da Silva, L.P., Mata, V.A., Lopes, P.B., Pereira, P., Jarman, S.N., Lopes, R.J., and Beja, P., 2019. Advancing the integration of multi-markers metabarcoding data in dietary analysis of trophic generalists. Mol. Ecol. Resour., 19: 1420-1432. https://doi.org/10.1111/1755-0998.13060
De, B.M., Miquel, C., Boyer, F., Mercier, C., Rioux, D., Coissac, E., and Taberlet, P., 2014. DNA metabarcoding multiplexing and validation of data accuracy for diet assessment: Application to omnivorous diet. Mol. Ecol. Resour., 14: 306-323. https://doi.org/10.1111/1755-0998.12188
Fu, Y.Q., Jia, X.D., Hu, J.C., Guo, Y.S., Zhu, H.B., Liu, W.H., and Wang, Y.S., 2006. Summer habitat selection by sika deer in Taohongling Nature Reserve, Jiangxi Province. Sichuan J. Zool., 25: 863-865.
Gao, Y.M., Yu, B., Wang, J.Q., Wu, W.G., and Gao, B., 2009. Habitat characteristics used by Sika deer at taohongling Natural Reserve. Jiangxi Sci., 27: 877-878.
Goldberg, A.R., Conway, C.J., Tank, D.C., Andrews, K.R., Gour, D.S., and Waits, L.P., 2020. Diet of a rare herbivore based on dna metabarcoding of feces: selection, seasonality, and survival. Ecol. Evol., 10: 7627-7643. https://doi.org/10.1002/ece3.6488
Guo, Y.P., Zhang H., Zhao X.G., Luo H.L., and Zhang Y.J., 2021. Applications of DNA Metabarcoding in diet identification of herbivores. Biotechnol. Bull., 37: 252-260.
Guo, Y.S., 2000. Study on the food habits of Sichuan Sika Deer (Cervus nippon sichuanicus). J. Sichuan Teach. Coll. (natl. Sci.), 22: 112-119.
Guo, Y.S., and Zheng, H.Z., 2001. On the geological distribution taxonomic status of species and evolutionary history of sika deer in China. Acta Theriol. Sin., 20: 168-179.
He, Y.Y., Liu, K.L., Dong, J.J., Yu, Y.F., and He, K., 2019. Comparision of vigilance-related calls between Cervus nippon kopschi and Cervus nippon hortulorum. Sichuan J. Zool., 38: 241-248.
Heroldova, M., Cizmar, D., and Tkadlec, E., 2010. Predicting rodent impact in crop fields by near-infrared reflectance spectroscopy analysis of their diet preferences. Crop Prot.., 29: 773-776. https://doi.org/10.1016/j.cropro.2010.02.009
Holechek, J.L., Gross, B., Dabo, S.M., and Stephenson, T., 1982. Effects of sample preparation, growth stage, and observer on microhistological analysis of herbivore diets. J. Wildl. Manag., 46: 502-505. https://doi.org/10.2307/3808666
Hou, J.J., Li, L., Wang, Y.F., Wang, W.J., Zhan H.Y., Dai N.H., and Lu P., 2021. Influences of submerged plant collapse on diet composition, breadth, and overlap among four crane species at Poyang Lake, Front. Zool., 18: https://doi.org/10.1186/s12983-021-00411-2
Kaneko, H., and Lawler, I.R., 2006. Can near infrared spectroscopy be used to improve assessment of marine mammal diets via fecal analysis? Mar. Mammal. Sci., 22: 261-275. https://doi.org/10.1111/j.1748-7692.2006.00030.x
Levins, R., 1968. Evolution in changing environments. Princeton University Press, Princeton. https://doi.org/10.1515/9780691209418
Li, J.N., and Liu, J.K., 2003. Ecological implication and behavior mechanism of food selection of mammalian herbivores. Chinese J. appl. Ecol., 14: 439-442.
Li, F.W., Kuo, L.Y., Rothfels, C.J., Ebihara, A., Chiou, W.L., Windham, M.D., and Pryer, K.M., 2011. rbcL and matK earn two thumbs up as the core DNA barcode for ferns. PLoS One, 6: e26597. https://doi.org/10.1371/journal.pone.0026597
Liu, G., Ning, Y., Xia, X.F., and Gong, M.H., 2018. The application of high-throughput sequencing technologies to wildlife diet analysis. Acta Ecol. Sin., 38: 3347-3356. https://doi.org/10.5846/stxb201706151092
Lu, Q., Hu, Q., Shi, X.G., Jin, S.L., Li, S., and Yao, M., 2019. Metabarcoding diet analysis of snow leopards (Panthera uncia) in Wolong National Nature Reserve, Sichuan Province. Biodivers. Sci., 27: 960-969. https://doi.org/10.17520/biods.2019101
Magoc, T., and Salzberg, S.L., 2011. FLASH: fast length adjustment of short reads to improve genome assemblies. Bioinformatics, 27: 2957-2963. https://doi.org/10.1093/bioinformatics/btr507
Mallott, E.K., Garber, P.A., and Malhi, R.S., 2018. trnL outperforms rbcL as a DNA metabarcoding marker when compared with the observed plant component of the diet of wild white-faced capuchins (Cebus capucinus, Primates). PLoS One, 13: e0199556. https://doi.org/10.1371/journal.pone.0199556
Mata, V.A., Rebelo, H., Amorim, F., Mccracken, G.F., Jarman, S., and Beja, P., 2019. How much is enough? Effects of technical and biological replication on metabarcoding dietary analysis. Mol. Ecol., 28: 165-175. https://doi.org/10.1111/mec.14779
Nakahama, N., Furuta, T., Audo, H., and Setsuko, S., 2021. DNA meta-barcoding revealed that sika deer foraging strategies vary with season in a forest with degraded understory vegetation. For. Ecol. Manage., 484: 118637. https://doi.org/10.1016/j.foreco.2020.118637
Parameswaran, P., Jalili, R., Tao, L., Shokralla, S., Gharizadeh, B., Ronaghi, M., and Fire, A.Z., 2007. A pyrosequencing-tailored nucleotide barcode design unveils opportunities for large-scale sample multiplexing. Nucl. Acids Res., 35: e130. https://doi.org/10.1093/nar/gkm760
Pielou, E.C., 1969. An introduction to mathematical ecology. Wiley-Interscience, New York.
Shao, X.N., Song, D.Z., Huang, Q.W., Li, S., and Yao, M., 2019. Fast surveys and molecular diet analysis of carnivores based on fecal DNA and metabarcoding. Biodivers. Sci., 27: 543-556. https://doi.org/10.17520/biods.2018214
Smith, E.P., 1982. Niche breadth, resource availability, and inference. Ecology, 63: 1675–1681. https://doi.org/10.2307/1940109
Sun, P., Ling, J.Z., Zhang, H., Tang, B.J., and Jiang, Y.Z., 2021. Diet composition and feeding habits of black sea bream (Acanthopagrus schlegelii) in Xiangshan Bay based on high-throughput sequencing. Acta Ecol. Sin., 41: 1221-1228. https://doi.org/10.5846/stxb201911022308
Taberlet, P., Coissac, E., Pompanon, F., Gielly, L., Miquel, C., Valentini, A., Vermat, T., Corthier, G., Brochmann, C., and Willerslev, E., 2007. Power and limitations of the chloroplast trnL(UAA) intron for plant DNA barcoding. Nucl. Acids Res., 35: e14. https://doi.org/10.1093/nar/gkl938
Tercel, M.P.T.G., Symondson, W.O.C., and Cuff, J.P., 2021. The problem of omnivory: A synthesis on omnivory and dna metabarcoding. Mol. Ecol., 30: 2199-2206. https://doi.org/10.1111/mec.15903
Valentini, A., Pompanon, F., and Taberlet, P., 2009. DNA barcoding for ecologists. Trends Ecol. Evol., 24: 110-117. https://doi.org/10.1016/j.tree.2008.09.011
Vázquez-Baeza, Y., Pirrung, M., Gonzalez, A., and Knight, R., 2013. EMPeror: A tool for visualizing high-throughput microbial community data. Gigascience, 2: 16. https://doi.org/10.1186/2047-217X-2-16
Wang, L., 2018. The countermeasures and suggestions of improving the cervusnippon habitat in Jiangxi Taohongling National Nature Reserve. J. Hebei For. Sci. Technol., 1: 68-70.
Wang, S., 1998. China red data book of endangered animals: Mammalia. Science Press, Beijing (in Chinese).
Wang, X., Jiang, H.X., and Zhang, Y.N., 2015. Application of stable isotope analyses to avian diets and trophic structure. Acta Ecol. Sin., 35: 5556-5569. https://doi.org/10.5846/stxb201402120243
Wang, Z.Y., Meng, D.H., Luo, Y., Teng, L.W., Gao, H., Wang, J.F., and Liu, Z.S., 2019. Spring food-habits of red deer (Cervus elaphus alashanicus) in Helan Mountains, China. Chinese J. Wildl., 40: 825-831.
Wu, W.G., Zhu, W., Gao, Y.M., Wang, J.Q., and Wang, L.Z., 2012. Biodiversity in Jiangxi Taohong Ridge Sika Deer Nature Reserve. Chinese J. Wildl., 33: 221-224.
Xu, J., Bao, X., Liu, Z.S., Gao, H., Zhao, C., Sun, Y.J., Wang, J.F., and Teng, L.W., 2018. A comparative study of autumn diets of Alpine musk deer (Moschus chrysogaster) and Alashan red deer (Cervus alashanicus) in the Helan Mountains, China. Acta Ecol. Sin., 38: 1-7. https://doi.org/10.5846/stxb201703100404
Yang, Y.Z., 2016. Diet analysis and gut microbiota of herbivorous Anseriformes in the middle and lower Yangtze floodplain. PhD thesis, University of Science and Technology of China, Hefei, China.
Zhang, C.Y., Wu, J.P., and Huang, X.Y., 2011. Evaluation of scatological analysis of Sika deer food habits. Chinese J. Wildl., 32: 199-202.
Zhang, Q.J., Yang, B., Fu, Q., Wang, L., Gong, X., and Zhang, Y.B., 2020. The winter diet of sambar (Rusa unicolor) in the Qionglai Mountains. Biodivers. Sci., 28: 1192-1201. https://doi.org/10.17520/biods.2020063
Zheng, X.Q., Wang, Q., Huang, L.F., Wang, J.J., Lin, R.C., Huang, D.Y., and Sun, X.H., 2015. Feeding habits for two dominant amphipod species in the Yundang Lagoon based on stable carbon and nitrogen isotope analysis. Acta Ecol. Sin., 35: 7589-7597. https://doi.org/10.5846/stxb201404200779
Zhou, Y.X., Li, Y.K., Li, J.Q., Liu, W.H., Shao, R.Q., Zhong, Y.F., and Cao, K.Q., 2019. Use of camera trapping to investigate animal diversity in Taohongling Sika Deer National Nature Reserve. Acta Ecol. Sin., 39: 4975-4984. https://doi.org/10.5846/stxb201805251148
To share on other social networks, click on any share button. What are these?


