
Advances in Animal and Veterinary Sciences


Mini Review Article
Advances in Animal and Veterinary Sciences 2 (6): 312 – 315Metagenomics: a Novel Tool to Unravel the Secrets of Nature
Varuna Purushothama Panicker*, Anu Gopalakrishnan1
- College of veterinary and Animal Sciences, Mannuthy–680651 Thrissur, Kerala
*Corresponding author: drpanickerpvarunavet@gmail.com
ARTICLE CITATION:
Panicker VP, Gopalakrishnan A (2014). Metagenomics: a novel tool to unravel the secrets of nature. Adv. Anim. Vet. Sci. 2 (6): 312 – 315.
Received: 2014–06–08, Revised: 2014–06–24, Accepted: 2014–06–25
The electronic version of this article is the complete one and can be found online at
(
http://dx.doi.org/10.14737/journal.aavs/2014/2.6.312.315
)
which permits unrestricted use, distribution, and reproduction in any medium, provided the original work is properly cited
ABSTRACT
Metagenomics is the biotechnological tool which helps to know the genetic makeup of the wide variety of uncultivable microorganisms. This helps us to understand the diversity of the ecosystem. This genomic analysis tool provides information about the unknown biocatalysts and biomolecules produced by the microorganisms which may be helpful in various biotechnological approaches. This review also highlights various screening methods in metagenomics to unravel the genetic makeup of the uncultivable microorganisms. This will later help in the engineering of these newly reported biocatalysts and biomolecules for specific processes. The expansion of metagenomic techniques with the development of various sophisticated methods will offer the way to find out even more valuable information about the microbial world.
Prokaryotes represent the largest proportion of life forms on earth, which comprises 106 to 108 different genotypes (Sleator et al., 2008). These microorganisms are responsible for most of the chemical cycles on earth, which are essential for our existence. In addition, microorganisms serve human beings by maintaining our health, fermenting food and producing drugs (Backhed et al., 2005). Most of these microorganisms live in communities, many of those communities are complex with high magnitude of diversity with thousands of interacting members (DeLong, 2002) where they will compete for basic needs like space, air, etc. For the better understanding of life, it is essential to understand the diversity of these microorganisms in the community (Tyson et al., 2004). Most of the research on microorganisms are based on culturing organisms in the laboratory. Major difficulty encountered by researchers in the field of microbial study was, how to study those organisms which do not grow under standard culturing conditions (Handelsman, 2004).
The term metagenomics was first coined by Handelsman in 1998, for habitat based investigation of mixed microbial population at the DNA level (Steele and Streit, 2005). Metagenomics provides a culture–independent way to access unculturable microorganisms and is now possible to study the genome of all those organisms. Thus metagenomics revolutionized the field of microbiology which offers a window to understand previously unknown and uncultivable microorganisms.
Life on earth was flourished due to a transition from the anaerobic to aerobic forms of photosynthetic bacteria. Due to this transition oxygen began to accumulate in the atmosphere until it was sufficient to support the life of aerobic organisms. Once oxygen concentration reached at a very high concentration, oxygen molecule began to collide and produced ozone. Later this ozone gas accumulated in the stratosphere and protected the life forms on earth from ultraviolet light (Handelsman et al., 1998). Yet another group of microorganisms evolved are nitrogen fixers. These bacteria could break triple bonded nitrogen molecule and fix atmospheric nitrogen for the usage of terrestrial living beings. Human health is under constant check by human microbiome such as gut microflora, when the balance of gut microbial community is compromised, many diseases like colon cancer, inflammatory bowel disease, obesity and diabetes may occur. All these microbes coevolved with the human species, produces an intertwined web of dependency and communication (Turnbaugh et al., 2006). A large proportion of the drugs available today are synthesized from bacteria and fungi. The discovery of antibiotics has transformed human existence by providing an outstanding way for the treatment of infectious diseases. In addition microorganisms play an important role in providing industrial enzymes and polymers, cleaning up toxic waste products and can be employed in the process of fermentation etc.
The main practice in bacteriology for identifying microorganisms for years was by culturing. Culturing techniques brought a gap in our knowledge about the wide variety of microorganisms that cannot be cultured. In 1980s, before the advent of metagenomics, another method was available to assess the uncultured microorganisms. This was by the nucleotide sequence analysis of small subunit ribosomal ribonucleic acid genes (rRNA, 16Sr RNA for prokaryotes and 18S rRNA for eukaryotes) (Schmidt et al., 1991). Analysis of these signature sequences helped to generate awareness about the amazing diversity of the microbial community, but provided little insight into the
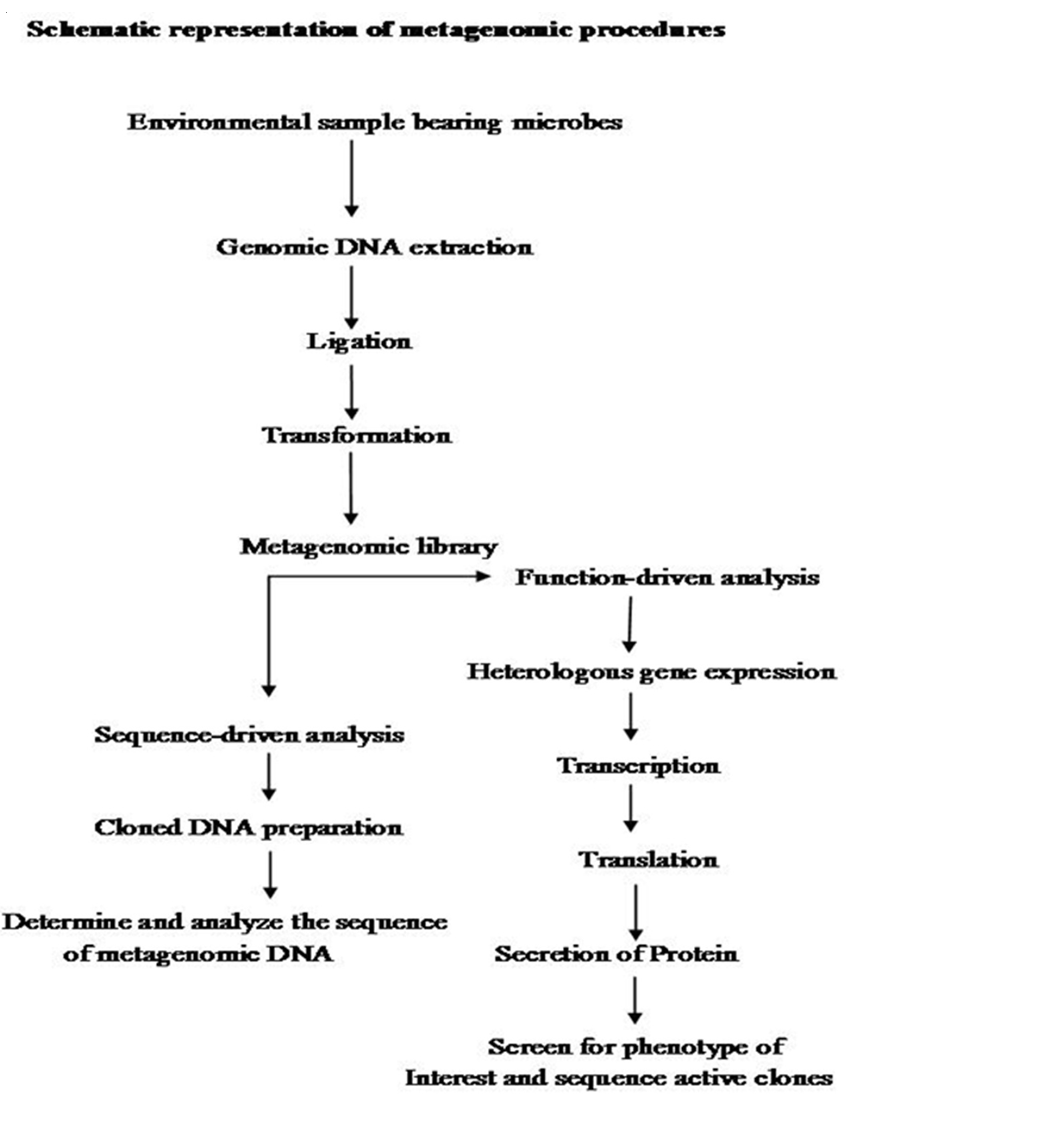
functional role of those organisms. Under these circumstances there was an urgent need among researchers for new tools to discover and analyze the function of these uncultivable microbes. This lead to the discovery of Metagenomics– the genomic analysis of microbial communities (Schneider and Riedel, 2010).
The first step in Metagenomics involves extraction of DNA directly from an environmental samples eg; soil, (Voget et al., 2006 and Waschkowitz et al., 2009) seawater (Stein et al., 1996), ground water (Uchiyama et al., 2005), antarctic desert soil (Heath et al., 2009), human microbiome etc. Microbial DNA isolation from extreme environment is still a technological challenge because many times the DNA extraction procedure standardized for mesophilic sample may not be applicable for the samples isolated from extreme conditions. Hence various methods have been developed for the isolation of high quality DNA from a variety of extreme environments like hot water spring (Hardeman and Sjoling, 2007). The extracted DNA contains a pool of genomes from different organisms. Second step is to insert these DNAs into a surrogate host, such as Escherichia coli. These inserted DNAs could then be studied either in sequence driven analysis or function based analysis (Pathak et al., 2009).
Metagenomics was adopted by the scientific community in the 1990s with various goals (Handelsman et al., 1998). It was the first and foremost method to learn about the functional contributions to the biosphere made by the members of the microbial community that could not be cultured. To map microbial communities that are associated with the human gut, mouth, skin and vagina, Human Microbiome Project was founded in 2009.
Metagenomics was designed with several practical gains, such as the discovery of new genes and gene products that would lead to agricultural innovations, medicinal chemistry and industrial processes (Handelsman, 2004., Riesenfeld et al., 2004). These goals were achieved by two parallel methodological approaches like sequence–driven metagenomics and function– driven metagenomics. In the sequence–driven metagenomics approach, the DNA from the environment of interest is sequenced and analyzed. This requires the designing of DNA probe or primers which are driven from conserved regions of already–known genes (Simon and Daniel, 2011). Metagenomics sequences are then compared to the sequence data available in publicly available database such as GENBANK. Sequenced genes can then be grouped into groups of similar predicted functions. This approach led to the successful identification of some novel enzymes like catalogs, DNA polymerases etc. (Knietsch et al., 2003).
In function– driven metagenomics, the DNA extracted from the environment is captured and stored in a surrogate host. Instead of sequencing, the scientists will screen the captured fragments of DNA for functional analysis. Isolation of genes encoding novel biomolecules are based on the metabolic activities of metagenomic–library–containing clones (Simon and Daniel, 2011). Novel biomolecules are recovered by three different approaches in function–driven metagenomics: phenotypical detection of the desired activity of microorganisms, heterologous complementation of host strains or mutants and by induced gene expression. Phenotypical detection is by using chemical dyes and chromophore–bearing derivatives of enzyme substrate incorporated into the growth medium, where they register the specific metabolic products produced by the individual clones (Simon and Daniel, 2009). A second category of function– driven metagenomic analysis is based on heterologous complementation of host strains or mutants of host strains which require the targeted gene for growth under selective conditions (Simon and Daniel, 2010). This approach is highly selective for the targeted genes. Third type of activity–driven screening termed as substrate–induced gene expression screening (SIGEX) (Uchiyama et al., 2005). This high–throughput screening method employs a GFP (Green Fluorescent Protein) expression vector in combination with fluorescence–activated cell sorting. Later on a similar type of screening method, designated as metabolite–regulated expression (MATREX) has been published to identify metagenomic clones producing small molecules like biosensors.
Even though these approaches are effective enough to inform us about the diversity of the microbial world they have limitations also. In sequence–driven metagenomics if a metagenomic gene does not show a sequence similarity to any of the gene of known function deposited in public database, then little can be learned about the gene or its gene product. In function– driven metagenomics the major drawback is that most genes from organisms in wild communities may not be expressed easily by a given surrogate host.
Later the development of next–generation sequencing techniques and other affordable methods allowing large–scale analysis of microbial communities resulted in novel applications in the field of metagenomics, such as community metagenomics, metatranscriptomics (Sorek and Cossart, 2010) and metaproteomics (Wilmes et al., 2008). Introduction of next–generation sequencing platforms, such as Genome Analyzer of Illumina (Bentley, 2006), Roche 454 sequencer and the SOLiD system of Applied Biosystems had a big impact on metagenomic research (Metzker, 2010). Increased sequencing efficiency and reduction in cost by way of these emerging techniques led to the increase in size and number of metagenomic sequencing projects.
Metagenomics is one of the fastest growing research field of biology with full of promises. It provides a window to the researcher which was unseen before. It promises to provide a more complete understanding of global biological cycles that keeps biosphere in balance, organism responsible for production of enzymes, proteins, antibiotics etc. The development of new improved methods of DNA isolation, cloning techniques and screening strategies allowed assessment and exploiting of microbes from extreme and inhospitable environments such as hot springs, glaciers, hypersaline basins etc. Advent of next–generation sequencing and advanced bioinformatics tools helped in analysis and comparison of the metagenomic data sets with respect to phylogenetic and metabolic diversity.
CONFLICT OF INTEREST
No conflict of interest among authors.
REFERENCES
Backhed F, Ley RF, Sonnenburg JL, Peterson DA, Gordon JI (2005). Host–bacterial in the human intestine. Sci. 307: 1915 – 1920.
http://dx.doi.org/10.1126/science.1104816
PMid:15790844
Bentley DR (2006). Whole–genome resequencing. Curr. Opin. Genet. Dev. 16: 545 – 552.
http://dx.doi.org/10.1016/j.gde.2006.10.009
PMid:17055251
DeLong EF (2002). Microbial population genomics and ecology. Curr. Opin. Microbiol. 5:520 - 524
http://dx.doi.org/10.1016/S1369-5274(02)00353-3
Handelsman J (2004). Metagenomics: application of genomics to uncultured microorganisms Micrbiol. Mol. Biol. Rev. 68: 669 – 685.
Handelsman J, Rondon MR, Brandy SF, Clardy J, Goodman RM (1998). Molecular biology provides access to the chemistry of unknown soil microbes: a new frontier for natural products. Chem. Biol. 5: 245 – 249.
http://dx.doi.org/10.1016/S1074-5521(98)90108-9
Hardeman F, Sjoling S (2007). Metagenomic approach for the isolation of a novel low–temperature–active lipase from uncultured bacteria of marine sediment. FEMS Microbiol. Ecol. 59: 524 – 534.
http://dx.doi.org/10.1111/j.1574-6941.2006.00206.x
PMid:17328767
Heath C, Hu XP, Cary SC, Cowan D (2009). Identification of a novel alkaliphilic esterase active at low temperatures by screening a metagenomic library from Antarctic desert soil. Appl. Environ. Microbiol. 75: 4657 – 4659.
http://dx.doi.org/10.1128/AEM.02597-08
PMid:19411411 PMCid:PMC2704825
Knietsch A, Bowien S, Whited G, Gottschalk G, Daniel, R (2003). Identification and characterization of coenzyme B12– dependent glycerol dehydratase and diol dehydratase– encoding genes from metagenomic DNA libraries derived from enrichment cultures. Appl. Environ. Microbiol. 69: 3048 – 3060.
http://dx.doi.org/10.1128/AEM.69.6.3048-3060.2003
PMid:12788698 PMCid:PMC161467
Metzker ML (2010). Sequencing technologies– the next generation. Nature Rev. Genet. 11: 31 – 46.
http://dx.doi.org/10.1038/nrg2626
PMid:19997069
Pathak GP, Ehrenreich A, Losi A, Streit WR, Gartner W (2009). Novel blue light–sensitive proteins from a metagenomic approach. Environ. Microbiol. 11: 2388 – 2399.
http://dx.doi.org/10.1111/j.1462-2920.2009.01967.x
PMid:19538504
Riesenfeld CS, Schloss PD, Handelsman J (2004). Metagenomics: genomic analysis of microbial communities. Annu. Rev. Genet. 38: 525 – 552.
http://dx.doi.org/10.1146/annurev.genet.38.072902.091216
PMid:15568985
Schmidt TM, DeLong EF, Pace NR (1991). Analysis of a marine picoplankton community by 16S rRNA gene cloning and sequencing. J. Bacteriol. 173: 4371 – 4378.
PMid:2066334 PMCid:PMC208098
Schneider T, Riedel K (2010). Environmental proteomics: analysis of structure and function of microbial communities. Proteomics. 10: 785 – 798.
http://dx.doi.org/10.1002/pmic.200900450
PMid:19953545
Simon C, Daniel R (2009). Achievements and new knowledge unraveled by metagenomic approaches. Appl. Microbiol. Biotechnol. 85: 265 – 276.
http://dx.doi.org/10.1007/s00253-009-2233-z
PMid:19760178 PMCid:PMC2773367
Simon C, Daniel R (2010). Construction of small–insert and large insert metagenomic libraries. Methods Mol. Biol. 668: 39 – 50.
http://dx.doi.org/10.1007/978-1-60761-823-2_2
PMid:20830554
Simon C, Daniel R (2011). Metagenomics analysis: Past and future trends. Appl. Environ. Microbiol. 77: 1153 – 1161.
http://dx.doi.org/10.1128/AEM.02345-10
PMid:21169428 PMCid:PMC3067235
Sleator RD, Shortall C, Hill C (2008). Metagenomics. Lett. Appl. Microbiol. 47:361 – 366.
http://dx.doi.org/10.1111/j.1472-765X.2008.02444.x
PMid:19146522
Sorek R, Cossart P (2010). Prokaryotic transcriptomics: a new view on regulation, physiology and pathogenesity. Nat. Rev. Genet. 11: 9–16.
http://dx.doi.org/10.1038/nrg2695
PMid:19935729
Stein JL, Marsh TL, Wu KY, Shizuya H, DeLong EF (1996). Characterization of uncultivated prokaryotes: isolation and analysis of a 40–kilobase–pair genome fragment from a planktonic marine archeon. J. Bacteriol. 178: 591 – 599.
PMid:8550487 PMCid:PMC177699
Turnbaugh PJ, Ley RE, Mahowald MA, Magrini V, Mardis ER, Gordon JI. (2006). An obesity–associated gut microbiome with increased capacity for energy harvest. Nature. 444: 1027 – 1031.
http://dx.doi.org/10.1038/nature05414
PMid:17183312
Tyson GW, Chapman J, Hugenhiltz P, Allen EE, Ram RJ, Richardson PM, Solovyev VV, Rubin EM, Rokhsar DS, Banfield JF (2004). Community structure and the metabolism through reconstruction of microbial genomes from the environment. Nature. 428: 37 – 43.
http://dx.doi.org/10.1038/nature02340
PMid:14961025
Uchiyama T, Abe T, Ikemura T, Watanable K (2005). Substrate induced gene–expression screening of environmental metagenome libraries for isolation of catabolic genes. Nat. Biotechnol. 23: 88 – 93.
http://dx.doi.org/10.1038/nbt1048
PMid:15608629
Voget S, Steele HL, Streit WR (2006). Characterization of a metagenome–derived halotolerant cellulose. J. Biotechnol. 126: 26 – 36.
http://dx.doi.org/10.1016/j.jbiotec.2006.02.011
PMid:16584799
Waschkowitz T, Rockstroh S, Daniel R (2009). Isolation and characterization of metalloproteases with a novel domain structure by construction and screening of metagenomic libraries. Appl. Environ. Microbiol. 75: 2506 – 2516.
http://dx.doi.org/10.1128/AEM.02136-08
PMid:19218412 PMCid:PMC2675231
Wilmes P, Wexler M, Bond PL (2008). Metaproteomics provides functional insight into activated sludge wastewater treatment. PLoS One. 3: e1778.

