

Cytotoxic Effect of MTH1 Gene Silencing in Gemcitabine Resistant Breast Cancer Cells





Cytotoxic Effect of MTH1 Gene Silencing in Gemcitabine Resistant Breast Cancer Cells
Mahak Fatima1, Memoona Syed1, Rabia Zeeshan2, Farah Rauf Shakoori3, Naveed Shahzad4, Moazzam Ali1 and Zeeshan Mutahir1*
1Institute of Biochemistry and Biotechnology, University of the Punjab, 54590 Lahore, Pakistan
2Interdisciplinary Research Centre in Biomedical Materials, COMSATS University Islamabad, Lahore Campus, 54000 Lahore, Pakistan
3Department of Zoology, University of the Punjab, 54590 Lahore, Pakistan
4School of Biological Sciences, University of the Punjab, 54590 Lahore, Pakistan
ABSTRACT
Nudix hydrolase 1 (NUDT1) or human MutT homolog 1 (MTH1) catalyzes the conversion of oxidized purine nucleotides to their monophosphate forms, thus preventing their incorporation into DNA and subsequently to oxidative damage. The present study explores the relationship between MTH1 gene silencing and its outcome on the growth of gemcitabine resistant breast cancer cells named MCF7-R. For this purpose, we transfected MCF7-R cells with MTH1 specific validated siRNA. Successful transfection of siRNA and consequent cytotoxicity in MCF7-R cells was confirmed by qRT-PCR, western blotting and cell viability assay. As a result of siRNA transfection, MTH1 protein was knockdown to approximately 77% in the transfected sample and resulted in a 1.75-fold increase in sensitivity of MCF7-R cells to gemcitabine. Moreover, higher expression of p21 protein was also observed in transfected MCF7-R cells that may indicate induced cell death. This study highlights the effect of MTH1 gene silencing in drug-resistant cancer cells as a mean to improve combined therapies for targeting drug-resistant cancers.
Article Information
Received 06 April 2020
Revised 05 May 2020
Accepted 11 May 2020
Available online 04 September 2020
Authors’ Contribution
MF and MS performed the experiments and contributed in writing the manuscript. RZ, FRS and NS contributed in writing the manuscript. MA helped in experimental work. ZM planned the study, analyzed the results and contributed in writing the manuscript.
Key words
Breast cancer, Gemcitabine, MTH1, ROS, siRNA, Oxidative stress
DOI: https://dx.doi.org/10.17582/journal.pjz/20200406140416
* Corresponding author: zeeshan.ibb@pu.edu.pk
0030-9923/2020/0006-2145 $ 9.00/0
Copyright 2020 Zoological Society of Pakistan
INTRODUCTION
Reactive oxygen species (ROS) are highly charged molecules that are produced within cells under normal physiological conditions or by exposure to certain chemicals and ionizing radiations (Ames et al., 1993). Once produced, ROS can interact with cellular components, including nucleic acids, lipids and proteins, thus causing oxidative damage and undermining genomic integrity (Ames et al., 1993; Cooke et al., 2003; Reuter et al., 2010; Winyard et al., 2011). ROS may oxidize nucleotides and the incorporation of oxidized nucleotides into DNA can directly damage DNA, thus enhancing mutagenesis and onset of different diseases like cardiovascular diseases, neurodegeneration and cancer (Ames et al., 1993; Cooke et al., 2003). Among all damaging molecules, oxidized guanine (8-Oxo-G) and deoxyguanosine (8-Oxo-dG) serve as notable source for spontaneous mutations by incorporating into nucleic acids. 8-Oxo-dGTP is produced by oxidation of deoxyguanosine triphosphate (dGTP) which readily pairs with adenine or cytosine, causing transversion mutations (Maki and Sekiguchi, 1992; Sheng et al., 2012; Tajiri et al., 1995). Accumulation of spontaneous mutations in the genome can disrupt its stability, hence linked with the onset of cancer by provoking rapid proliferation and inflammation (Nakabeppu et al., 2006). To combat the effect of ROS induced oxidative stress, there are various DNA mechanisms that operate in organisms, including certain enzymes such as MutT of E. coli and its human homolog MTH1 (Maki and Sekiguchi, 1992; Sakumi et al., 1993). MTH1 is also known as Nudix hydrolase 1 or NUDT1. It is an 18 KDa phosphohydrolase which hydrolyses the conversion of oxidized purines such as 8-Oxo-dGTP and 2-OH-dATP to their monophosphate form, thus impeding their incorporation into DNA and subsequent mutagenesis (Mo et al., 1992; Sakumi et al., 1993; Yoshimura et al., 2003). Disruption of MTH1 gene in a mouse model resulted in the accumulation of spontaneous mutations and tumorigenesis, which highlighted it as an indispensable enzyme for sanitization of oxidative nucleotides (Tsuzuki et al., 2001). MTH1 protein, which is not crucial for the survival of normal cells, has been proved inevitable for survival of cancer cells (Gad et al., 2014; Huber et al., 2014; Patel et al., 2015; Tu et al., 2016). Tumour cells undergo more rapid proliferation accompanied by a higher rate of metabolic processes, therefore, their DNA is more prone to oxidative damage (Freudenthal et al., 2015). To circumvent oxidative damage to DNA, cancer cells more strongly rely on MTH1 protein for sanitizing its nucleotide pool, as it has been shown by overexpression of MTH1 in several tumours and cancer cell lines compared to their normal counterparts (Speina et al., 2005). Furthermore, development of pharmacological tools and gene silencing techniques such as siRNA, shRNA and CRISPR based targeted approaches against MTH1 has been implemented to knock down its expression and proved effective for reducing cell viability in cancer cell lines (Gad et al., 2014; Huber et al., 2014; Kawamura et al., 2016; Kettle et al., 2016; Warpman Berglund et al., 2016). To the best of our knowledge, no study has been published so far which describes the effect of MTH1 gene silencing on the drug-resistant phenotype of cancer cells. The present study was aimed at exploring the effect of MTH1 gene silencing in gemcitabine resistant invasive breast carcinoma cell line (named MCF7-R) and to study its potential use in combined therapies for targeting cancer. This study highlights the cytotoxic effect of MTH1 gene silencing in gemcitabine resistant breast cancer cells in vitro.
MATERIALS AND METHODS
Chemicals
Gemcitabine, MTT (methylthiazolyldiphenyl-tetrazolium bromide), Isopropanol and Hydrochloric acid (HCL) were purchased from Sigma-Aldrich USA. MCF7 cells were grown in Dulbecco’s Modified Eagle Medium (DMEM) supplemented with Glutamax and Sodium Pyruvate (Catalog No. 1880272) and Opti-MEM™ (Catalog No. 1758537) were purchased from Gibco, USA. GeneJet RNA purification kit (K0731), Revertaid cDNA synthesis kit (K1622), FBS, penicillin/streptomycin, trypsin and PBS were purchased from Thermo Fisher Scientific USA, and validated MTH1 specific siRNA (s9030) was purchased from Ambion, USA. Goat anti-mouse IgG, horseradish peroxidase conjugate (Catalog No. G21040) and goat anti-rabbit IgG, horseradish peroxidase conjugate (Catalog No. G21234) were from Invitrogen, USA. Antibodies for β-actin (Catalog No sc-130656), p21 (Catalog No. sc-756) and MTH1 protein were purchased from Santa Cruz Biotechnology, USA. Lipofectamine® 3000 Transfection Reagent (Catalog number (L3000008) was from Invitrogen, USA.
Cell culture
2µM gemcitabine resistant breast cancer cell line MCF7-R was used in this study. The drug-resistant cancer cell line (MCF7-R) was generated earlier in our laboratory by gradually exposing the wild type MCF7 to increasing concentration of gemcitabine for seven months. MCF7-R cells were cultured in DMEM (supplemented with Glutamax and sodium pyruvate), to which FBS (10% v/v) and 100U/ml penicillin/streptomycin were also added. Gemcitabine was added to a final concentration of 2µM during culturing and subculturing of MCF7-R cells. Cells were grown in T-25 cell culture flasks with vented caps and kept in a humidified incubator with 5% CO2 and at 37˚C.
MTH1 gene silencing in MCF7-R cells
To identify the effect of MTH1 gene suppression in MCF7-R cells, we used validated MTH1 specific siRNA. The sequence of sense and antisense strands of MTH1 specific siRNA used in the current study were as follows, Sense sequence; 5’ CAUCUGGAAUUACUGGAUTT 3’, Anti-sense sequence; 5’ AUCCAGUUAAUUCCAGAUGAA 3’.
For transfection, MCF7-R cells were grown up to 70-80% confluency in two vented T-25 flasks, one for positive transfection with MTH1 specific siRNA and the other as an experimental control where nuclease-free water was used. Transfection of MTH1 specific siRNA was performed using Lipofectamine 3000 reagent (Invitrogen) following the manufacturer’s instructions. Briefly, MTH1 specific siRNA (150 pmol) and lipofectamine® 3000 (15µl per flask) were separately diluted in Opti-MEM™ media and mixed. After incubation of 10 minutes at room temperature, the mixture was directly added to the flask containing MCF7-R cells (named +ve transfection group). For the control experiment, nuclease-free water was used instead of siRNA, mixed with lipofectamine and added to the flask containing MCF7-R cells as described earlier (the control group named –ve transfection group). After the transfection, both groups of MCF7-R cells were incubated overnight at 37˚C in a humidified incubator with 5% CO2. After 24-h post-transfection incubation, part of cells from both groups were seeded into 96 well plates for cell viability assay. The remaining cells were incubated for further 72-h in complete growth medium for total RNA and protein isolation for subsequent quantitative real-time PCR and Western blot analyses, respectively.
Cell viability assay
MCF7-R cells from +ve and –ve transfection groups were seeded in triplicate in 96 well plate with a seeding density of 5000 cells per well in 100µl of complete medium. After 24-h of incubation, cells were supplemented with 100µl of fresh medium with different concentrations of gemcitabine except for the control group. Gemcitabine treated cells were further incubated at 37˚C in an incubator for 72 h. After completion of the incubation period, 10µl of MTT reagent (5mg/ml) was added per well and the plates were incubated at 37˚C for the formation of formazan crystals (purple coloured product formed due to dehydrogenase activity of viable cells). Formazan crystals were dissolved in 100µl of acidified-isopropanol solution (0.04N HCl in isopropanol) and absorbance was recorded at 492nm using LT-4500 microplate reader (LabTech). IC50 was calculated and survival curves were generated and analysed for positive and negative transfection groups using a computer program called GraphPad Prism (GraphPad Prism version 7.00 for Windows, GraphPad Software, La Jolla California USA, www.graphpad.com). IC50 value is the concentration of a drug or compound at which cells growth is inhibited to 50 percent. IC50 of MTH1 specific siRNA transfected and control groups was calculated according to equation 1 and folds of sensitivity were calculated by comparing the IC50 of both groups.
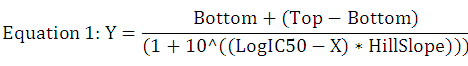
Where; X, log of dose or concentration; Y, response, decreases as X increases; Top and Bottom, Plateaus for the curve; HillSlope, slope factor.
Quantitative Real-time PCR for MTH1 expression
After transfection with MTH1 specific siRNA, total RNA was isolated from both +ve and –ve transfection groups separately, using a standard protocol of GeneJet RNA purification kit. RNA was quantified using NanoDrop™ 2000/2000c spectrophotometer (Thermo Fisher Scientific) and an equivalent amount of 1µg mRNA was used for DNase I treatment and subsequent cDNA synthesis according to the described protocol of RevertAid first-strand cDNA synthesis kit. The resultant cDNA was used to quantify the expression of MTH1 gene through quantitative real-time PCR (qRT-PCR) analysis using thermal cycler (BioRad CFX96). The gene-specific forward and reverse primers (200nM each) and SYBR Green qPCR master mix (Thermo Scientific) in a total reaction volume of 25µl were used in PCR reaction. The thermal profile of the reaction was as follows: initial denaturation at 95ºC for 15 min followed by 35 cycles at 95°C for 15 seconds, 58ºC for 20 seconds and 72ºC for 20 seconds. B2M gene expression was used as a normalisation control. Following were the sequences for gene-specific primers used in this study;
MTH1 forward primer; 5’CGACAGCTACTGGTTTCCAC3’,
MTH1 reverse primer; 5’GAGTGTGTAGTCCAGGATGG3’
B2M forward primer 5’TGCTGTCTCCATGTTTGATGTATCT3’
B2M reverse primer 5’ TCTCTGCTCCCCACCTCTAAGT 3’.
Western blot analysis
Total protein was isolated from both +ve and –ve transfected groups of MCF7-R cells using Laemmli’s method (Laemmli, 1970). The total protein content (number of cells equivalent used per lane) for each group was subjected to 4-12% SDS-PAGE for separation and transferred to PVDF membranes using wet transfer apparatus (Cleaver England) for overnight at 16V. PVDF membranes were then stained with Ponceau-S stain for confirmation of successful transfer and washed with distilled water. Membranes were blocked with skimmed milk (5% skimmed milk in TBST buffer) for 1-hour followed by incubation with primary antibodies against MTH1 (1:250, Santa Cruz), p21 (1:500, Santa Cruz), and β-actin (1:500, Santa Cruz). The membranes were washed with TBST for three times, 5 minutes each and incubated for an hour with secondary antibodies (HRP-conjugated). After rinsing the membranes with TBST, the proteins were incubated with HRP substrate and protein of interest were identified by chemiluminescence. Visualization of proteins was performed by developing an impression on Kodak films.
Statistical analysis
Data from each experiment were analyzed for statistical interpretation using computer programs; GraphPad Prism 7 and SPSS version 23. Data were considered statistically significant with a p-value less than 0.05.
RESULTS
MTH1 expression after MTH1-specific siRNA transfection
The quantitative real-time PCR assay was performed using SYBR Green-based chemistry for quantification of MTH1 gene expression from both +ve and –ve transfected groups of MCF7-R cells. Significant difference (p-value <0.0001) in the level of MTH1 gene expression was observed in +ve and –ve transfection groups (Fig. 1a). The expression of MTH1 protein was confirmed by western blot analysis. As a result of MTH1 specific siRNA transfection, the expression of MTH1 protein was found to be reduced to approximately 77% in +ve transfection group when compared to –ve transfection group (Fig. 1b). The results show successful transfection and silencing of MTH1 both at gene and protein level in +ve transfected cells.
Effect of MTH1 gene silencing on the viability of MCF7-R cell line
To study the effect of MTH1 knockdown on the viability of MCF7-R cells, MTT assay was performed using MCF7-R cells from +ve and –ve transfection groups. Cell viability was calculated for each group and survival curves were generated using GraphPad Prism 7 (Fig. 2). Nonlinear regression analysis was used to study the survival curves and computing IC50 for each group; IC50 was found to be 0.783±0.646µM for siRNA +ve transfected group and 1.370±0.0009µM for –ve transfected group. The ratio of IC50 of siRNA +ve and -ve transfection groups were compared and an increase of 1.75-fold in the sensitivity of resistant cells towards gemcitabine was observed in +ve transfection group after MTH1 gene silencing. A paired t-test was performed for statistical comparison of data obtained from +ve and –ve transfected groups and found highly significant with p-value of less than 0.05.
Detection of p21 protein in siRNA transfected cells
Inhibition of essential genes in cancer cell lines enhances the expression of certain proteins which serve as markers for the cell death process. p21 is a marker for cell death induction which acts either alone or with p53 protein in cell death activation pathways (Chen et al., 1996; Cho et al., 2011; Sheikh et al., 1996). To confirm the effect of MTH1 gene silencing on MCF7-R cells, western blot analysis for p21 protein was performed using p21 specific primary and secondary antibodies. The results showed significantly higher expression of p21 protein in siRNA transfected MCF7-R cells compared to the control group, which may indicate the induction of apoptosis or cell death as a result of silencing of MTH1 gene (Fig. 3). The expression of β-actin protein is used as an experimental control.
DISCUSSION
Over the past decade, MTH1 mechanism and function have been studied with respect to its molecular biology and effect on the growth of both normal and cancer cells (Coskun et al., 2015; Kennedy et al., 2003). It has been presented as a critical factor in cancer survival against ROS induced damage (Haghdoost et al., 2006; Speina et al., 2005). Several researchers emphasized the therapeutic efficacy of MTH1 inhibition in cancer cells by either designing active pharmacological inhibitors or interference based targeted approaches (Cho et al., 2011; Gad et al., 2014; Huber et al., 2014; Patel et al., 2015). However, the significance of targeting the MTH1 gene in drug-resistant cells has not been studied so far. Therefore, we used a validated siRNA against MTH1 gene in gemcitabine resistant MCF7 cells (MCF7-R).
Following knockdown of MTH1 gene using validated siRNA, the expression of MTH1 protein was reduced to approximately 77% in +ve transfected samples as shown in Figure 1. Our results are in accordance with earlier studies, where siRNA was used to abolish the expression of MTH1 and showed growth arrest in transfected samples (Lawless et al., 2010; Patel et al., 2015). To confirm the effect of MTH1 protein deficiency on gemcitabine resistant cancer cells, we performed cell proliferation assay for both +ve and -ve transfected samples. It is evident by our data that MTH1 downregulation affected the cell proliferation ability of MCF7-R cells together with 1.75-fold increase in sensitivity to gemcitabine (Fig. 2).
We further examined the expression of p21 protein as an apoptosis marker in both +ve and -ve transfected samples of MCF7-R cells and observed notably high p21 protein expression in +ve transfected group. These results are consistent with already reported studies, where MTH1 deficiency was correlated with the induction of ROS induced damage and cell death (Gad et al., 2014; Huber et al., 2014; Kettle et al., 2016; Rai et al., 2009). However, our results are contradictory to a recent study in which MTH1 deficiency in NSCLC cell lines was associated to induce genetic instability, but seemed unsuccessful to induce apoptosis (Abbas et al., 2018). A possible explanation for this contradiction is perhaps different genetic makeup and associated underlying pathways in different types of cancers.
CONCLUSION
In agreement to other studies, we believe that the use of MTH1 inhibitors alone in patients may not prove an effective therapeutic strategy due to upsurge in mutational pool associated with MTH1 inhibition which may further progress the disease. Therefore, we propose further studies aiming at understanding the underlying mechanism and interplay between drug resistance pathways and role of MTH1 protein to find more suitable inhibitor(s) as a potential therapeutic tool for the treatment of drug-resistant cancer cells.
ACKNOWLEDGEMENTS
A research grant to Dr. Zeeshan Mutahir by University of the Punjab is gratefully acknowledged to support this study. We are also thankful to Maryam Yousaf for her help in Western blot analysis.
Statement of conflict of interest
The authors have declared no conflict of interest.
REFERENCES
Abbas, H.H.K., Alhamoudi, K.M.H., Evans, M.D., Jones, G.D.D. and Foster, S.S., 2018. MTH1 deficiency selectively increases non-cytotoxic oxidative DNA damage in lung cancer cells: more bad news than good? BMC Cancer, 18: 423. https://doi.org/10.1186/s12885-018-4332-7
Ames, B.N., Shigenaga, M.K. and Gold, L.S., 1993. DNA lesions, inducible DNA repair, and cell division: three key factors in mutagenesis and carcinogenesis. Environ. Hlth. Perspect., 101: 35-44. https://doi.org/10.1289/ehp.93101s535
Chen, Y.C., Kuo, T.C., Lin-Shiau, S.Y. and Lin, J.K., 1996. Induction of HSP70 gene expression by modulation of Ca(+2) ion and cellular p53 protein by curcumin in colorectal carcinoma cells. Mol. Carcinog., 17: 224-234. https://doi.org/10.1002/(SICI)1098-2744(199612)17:4<224::AID-MC6>3.0.CO;2-D
Cho, W.C., Chow, A.S. and Au, J.S., 2011. MiR-145 inhibits cell proliferation of human lung adenocarcinoma by targeting EGFR and NUDT1. RNA Biol., 8: 125-131. https://doi.org/10.4161/rna.8.1.14259
Cooke, M.S., Evans, M.D., Dizdaroglu, M. and Lunec, J., 2003. Oxidative DNA damage: Mechanisms, mutation, and disease. FASEB J., 17: 1195-1214. https://doi.org/10.1096/fj.02-0752rev
Coskun, E., Jaruga, P., Jemth, A.S., Loseva, O., Scanlan, L.D., Tona, A., Lowenthal, M.S., Helleday, T. and Dizdaroglu, M., 2015. Addiction to MTH1 protein results in intense expression in human breast cancer tissue as measured by liquid chromatography-isotope-dilution tandem mass spectrometry. DNA Repair (Amst), 33: 101-110. https://doi.org/10.1016/j.dnarep.2015.05.008
Freudenthal, B.D., Beard, W.A., Perera, L., Shock, D.D., Kim, T., Schlick, T. and Wilson, S.H., 2015. Uncovering the polymerase-induced cytotoxicity of an oxidized nucleotide. Nature, 517: 635-639. https://doi.org/10.1038/nature13886
Gad, H., Koolmeister, T., Jemth, A.S., Eshtad, S., Jacques, S.A., Strom, C.E., Svensson, L.M., Schultz, N., Lundback, T., Einarsdottir, B.O., Saleh, A., Gokturk, C., Baranczewski, P., Svensson, R., Berntsson, R.P., Gustafsson, R., Stromberg, K., Sanjiv, K., Jacques-Cordonnier, M.C., Desroses, M., Gustavsson, A.L., Olofsson, R., Johansson, F., Homan, E.J., Loseva, O., Brautigam, L., Johansson, L., Hoglund, A., Hagenkort, A., Pham, T., Altun, M., Gaugaz, F.Z., Vikingsson, S., Evers, B., Henriksson, M., Vallin, K.S., Wallner, O.A., Hammarstrom, L.G., Wiita, E., Almlof, I., Kalderen, C., Axelsson, H., Djureinovic, T., Puigvert, J.C., Haggblad, M., Jeppsson, F., Martens, U., Lundin, C., Lundgren, B., Granelli, I., Jensen, A.J., Artursson, P., Nilsson, J.A., Stenmark, P., Scobie, M., Berglund, U. W. and Helleday, T., 2014. MTH1 inhibition eradicates cancer by preventing sanitation of the dNTP pool. Nature, 508: 215-221. https://doi.org/10.1038/nature13181
Haghdoost, S., Sjolander, L., Czene, S. and Harms-Ringdahl, M., 2006. The nucleotide pool is a significant target for oxidative stress. Free Radic. Biol. Med., 41: 620-626. https://doi.org/10.1016/j.freeradbiomed.2006.05.003
Huber, K.V., Salah, E., Radic, B., Gridling, M., Elkins, J.M., Stukalov, A., Jemth, A.S., Gokturk, C., Sanjiv, K., Stromberg, K., Pham, T., Berglund, U.W., Colinge, J., Bennett, K.L., Loizou, J.I., Helleday, T., Knapp, S. and Superti-Furga, G., 2014. Stereospecific targeting of MTH1 by (S)-crizotinib as an anticancer strategy. Nature, 508: 222-227. https://doi.org/10.1038/nature13194
Kawamura, T., Kawatani, M., Muroi, M., Kondoh, Y., Futamura, Y., Aono, H., Tanaka, M., Honda, K. and Osada, H., 2016. Proteomic profiling of small-molecule inhibitors reveals dispensability of MTH1 for cancer cell survival. Sci. Rep., 6: 26521. https://doi.org/10.1038/srep26521
Kennedy, C.H., Pass, H.I. and Mitchell, J.B., 2003. Expression of human MutT homologue (hMTH1) protein in primary non-small-cell lung carcinomas and histologically normal surrounding tissue. Free Radic. Biol. Med., 34: 1447-1457. https://doi.org/10.1016/S0891-5849(03)00176-X
Kettle, J.G., Alwan, H., Bista, M., Breed, J., Davies, N.L., Eckersley, K., Fillery, S., Foote, K.M., Goodwin, L., Jones, D.R., Kack, H., Lau, A., Nissink, J.W., Read, J., Scott, J.S., Taylor, B., Walker, G., Wissler, L. and Wylot, M., 2016. Potent and selective inhibitors of MTH1 probe its role in cancer cell survival. J. Med. Chem., 59: 2346-2361. https://doi.org/10.1021/acs.jmedchem.5b01760
Laemmli, U.K., 1970. Cleavage of structural proteins during the assembly of the head of bacteriophage T4. Nature, 227: 680-685. https://doi.org/10.1038/227680a0
Lawless, C., Wang, C., Jurk, D., Merz, A., Zglinicki, T. and Passos, J.F., 2010. Quantitative assessment of markers for cell senescence. Exp. Gerontol., 45: 772-778. https://doi.org/10.1016/j.exger.2010.01.018
Maki, H. and Sekiguchi, M., 1992. MutT protein specifically hydrolyses a potent mutagenic substrate for DNA synthesis. Nature, 355: 273-275. https://doi.org/10.1038/355273a0
Mo, J.Y., Maki, H. and Sekiguchi, M., 1992. Hydrolytic elimination of a mutagenic nucleotide, 8-oxodGTP, by human 18-kilodalton protein: sanitization of nucleotide pool. Proc. natl. Acad. Sci. U. S. A., 89: 11021-11025. https://doi.org/10.1073/pnas.89.22.11021
Nakabeppu, Y., Sakumi, K., Sakamoto, K., Tsuchimoto, D., Tsuzuki, T. and Nakatsu, Y., 2006. Mutagenesis and carcinogenesis caused by the oxidation of nucleic acids. Biol. Chem., 387: 373-379. https://doi.org/10.1515/BC.2006.050
Patel, A., Burton, D.G., Halvorsen, K., Balkan, W., Reiner, T., Perez-Stable, C., Cohen, A., Munoz, A., Giribaldi, M.G., Singh, S., Robbins, D.J., Nguyen, D.M. and Rai, P., 2015. MutT Homolog 1 (MTH1) maintains multiple KRAS-driven pro-malignant pathways. Oncogene, 34: 2586-2596. https://doi.org/10.1038/onc.2014.195
Rai, P., Onder, T.T., Young, J.J., McFaline, J.L., Pang, B., Dedon, P.C. and Weinberg, R.A., 2009. Continuous elimination of oxidized nucleotides is necessary to prevent rapid onset of cellular senescence. Proc. natl. Acad. Sci. U. S. A., 106: 169-174. https://doi.org/10.1073/pnas.0809834106
Reuter, S., Gupta, S.C., Chaturvedi, M.M. and Aggarwal, B.B., 2010. Oxidative stress, inflammation, and cancer: how are they linked? Free Radic. Biol. Med., 49: 1603-1616. https://doi.org/10.1016/j.freeradbiomed.2010.09.006
Sakumi, K., Furuichi, M., Tsuzuki, T., Kakuma, T., Kawabata, S., Maki, H. and Sekiguchi, M., 1993. Cloning and expression of cDNA for a human enzyme that hydrolyzes 8-oxo-dGTP, a mutagenic substrate for DNA synthesis. J. biol. Chem., 268: 23524-23530.
Sheikh, M.S., Garcia, M., Zhan, Q., Liu, Y. and Fornace, A.J., Jr., 1996. Cell cycle-independent regulation of p21Waf1/Cip1 and retinoblastoma protein during okadaic acid-induced apoptosis is coupled with induction of Bax protein in human breast carcinoma cells. Cell Growth Differ., 7: 1599-1607.
Sheng, Z., Oka, S., Tsuchimoto, D., Abolhassani, N., Nomaru, H., Sakumi, K., Yamada, H. and Nakabeppu, Y., 2012. 8-Oxoguanine causes neurodegeneration during MUTYH-mediated DNA base excision repair. J. clin. Invest., 122: 4344-4361. https://doi.org/10.1172/JCI65053
Speina, E., Arczewska, K.D., Gackowski, D., Zielinska, M., Siomek, A., Kowalewski, J., Olinski, R., Tudek, B. and Kusmierek, J.T., 2005. Contribution of hMTH1 to the maintenance of 8-oxoguanine levels in lung DNA of non-small-cell lung cancer patients. J. natl. Cancer Inst., 97: 384-395. https://doi.org/10.1093/jnci/dji058
Tajiri, T., Maki, H. and Sekiguchi, M., 1995. Functional cooperation of MutT, MutM and MutY proteins in preventing mutations caused by spontaneous oxidation of guanine nucleotide in Escherichia coli. Mutat. Res., 336: 257-267. https://doi.org/10.1016/0921-8777(94)00062-B
Tsuzuki, T., Egashira, A., Igarashi, H., Iwakuma, T., Nakatsuru, Y., Tominaga, Y., Kawate, H., Nakao, K., Nakamura, K., Ide, F., Kura, S., Nakabeppu, Y., Katsuki, M., Ishikawa, T. and Sekiguchi, M., 2001. Spontaneous tumorigenesis in mice defective in the MTH1 gene encoding 8-oxo-dGTPase. Proc. natl. Acad. Sci. U. S. A., 98: 11456-11461. https://doi.org/10.1073/pnas.191086798
Tu, Y., Wang, Z., Wang, X., Yang, H., Zhang, P., Johnson, M., Liu, N., Liu, H., Jin, W., Zhang, Y. and Cui, D., 2016. Birth of MTH1 as a therapeutic target for glioblastoma: MTH1 is indispensable for gliomatumorigenesis. Am. J. Transl. Res., 8: 2803-2811.
Warpman Berglund, U., Sanjiv, K., Gad, H., Kalderen, C., Koolmeister, T., Pham, T., Gokturk, C., Jafari, R., Maddalo, G., Seashore-Ludlow, B., Chernobrovkin, A., Manoilov, A., Pateras, I.S., Rasti, A., Jemth, A.S., Almlof, I., Loseva, O., Visnes, T., Einarsdottir, B.O., Gaugaz, F.Z., Saleh, A., Platzack, B., Wallner, O.A., Vallin, K.S., Henriksson, M., Wakchaure, P., Borhade, S., Herr, P., Kallberg, Y., Baranczewski, P., Homan, E.J., Wiita, E., Nagpal, V., Meijer, T., Schipper, N., Rudd, S.G., Brautigam, L., Lindqvist, A., Filppula, A., Lee, T.C., Artursson, P., Nilsson, J.A., Gorgoulis, V.G., Lehtio, J., Zubarev, R.A., Scobie, M. and Helleday, T., 2016. Validation and development of MTH1 inhibitors for treatment of cancer. Annls Oncol., 27: 2275-2283. https://doi.org/10.1093/annonc/mdw429
Winyard, P.G., Ryan, B., Eggleton, P., Nissim, A., Taylor, E., Lo Faro, M.L., Burkholz, T., Szabo-Taylor, K. E., Fox, B., Viner, N., Haigh, R.C., Benjamin, N., Jones, A.M. and Whiteman, M., 2011. Measurement and meaning of markers of reactive species of oxygen, nitrogen and sulfur in healthy human subjects and patients with inflammatory joint disease. Biochem. Soc. Trans., 39: 1226-1232. https://doi.org/10.1042/BST0391226
Yoshimura, D., Sakumi, K., Ohno, M., Sakai, Y., Furuichi, M., Iwai, S. and Nakabeppu, Y., 2003. An oxidized purine nucleoside triphosphatase, MTH1, suppresses cell death caused by oxidative stress. J. biol. Chem., 278: 37965-37973. https://doi.org/10.1074/jbc.M306201200
To share on other social networks, click on any share button. What are these?


