
Advances in Animal and Veterinary Sciences


Research Article
PCR Amplification Protocol for GC Rich Protamine Gene from Chicken Testis cDNA
Sanjeev Kumar Sharma1, Chathathayil Muhammedali Shafeeque2, Jag Mohan1, Parappurath Abdul Azeez2, Ram Pratap Singh2
1Physiology and Reproduction Division, Central Avian Research Institute, Izatnagar 243122, Bareilly, India; 2Sálim Ali Centre for Ornithology and Natural History Anaikatty 641108, Coimbatore, India.
Abstract | Amplification of GC rich templates using PCR is usually difficult compared to non-GC-rich targets. GC rich regions produce complex inter and intra strand folding (hairpins and loops) due to higher hydrogen bonding with neighbouring cytosine and guanine. These secondary structures in DNA are resistant to melting and cause Taq DNA polymerases to stall; they also hamper primer annealing, resulting in incomplete or non-specific amplifications. The aim of this study was to develop an easy PCR protocol to amplify very high GC rich (88% in the coding region) protamine gene of chicken. The cDNA used for amplification was synthesised from testis RNA and PCR products were detected by agarose gel electrophoresis. Protamine amplicon successfully amplified only with Go Taq DNA polymerase in the presence of DMSO. Optimization of MgCl2 concentration, buffer strength, annealing temperature, DMSO and the DNA template concentration, were important in the PCR reaction resulting in successful amplification. In addition, PCR started directly at hot plate is also important to get rid of primer-dimer formation.
Keywords | PCR, GC rich gene, DMSO
Editor | Kuldeep Dhama, Indian Veterinary Research Institute, Uttar Pradesh, India.
Received | October 20, 2014; Revised | November 15, 2014; Accepted | November 18, 2014; Published | November 20, 2014
*Correspondence | Ram Pratap Singh, Sálim Ali Centre for Ornithology and Natural History Anaikatty, India; Email: [email protected]
Citation | Sharma SK, Shafeeque CM, Mohan J, Azeez PA, Singh RP (2014). PCR amplification protocol for GC rich Protamine Gene from chicken testis cDNA. Adv. Anim. Vet. Sci. 2 (11): 599-605.
DOI | http://dx.doi.org/10.14737/journal.aavs/2014/2.11.599.605
ISSN (Online | 2307-8316; ISSN (Print) | 2309-3331
Copyright © 2014 Sharma et al. This is an open access article distributed under the Creative Commons Attribution License, which permits unrestricted use, distribution, and reproduction in any medium, provided the original work is properly cited.
INTRODUCTION
Polymerase chain reaction (PCR) technique enables researchers to produce millions of copies of a specific DNA or RNA sequence within a very short time period, and thereby has become one of the widely used techniques in the diagnostics and molecular biology laboratories. However, amplification of GC rich templates using PCR is usually difficult compared to non-GC-rich targets (McDowell et al., 1998). GC rich regions produce complex inter and intra strand folding (hairpins and loops) due to increased hydrogen bonding with neighbouring cytosine and guanine (Musso et al., 2006). These secondary structures in DNA are resistant to melting and cause Taq DNA polymerases to stall and they also hampers primer annealing, resulting in incomplete or non-specific amplification (Shore and Paul, 2010; Hubé et al., 2005). As a result of this, several approaches have been tried to amplify GC rich templates by adding dimethyl sulfoxide (DMSO), glycerol, polyethylene glycol, formamide, betaine, 7-deaza-dGTP, and dUTP in the reaction mixture (Baskaran et al., 1996; Chakrabarti and Schutt, 2001; Henke et al., 1997; Kang et al., 2005; Musso et al., 2006; Mutter and Boynton, 1995; Pomp and Medrano, 1991; Sidhu et al., 1996; Sun et al., 1993; Turner and Jenkins, 1995; Weissensteiner and Lanchbury, 1996). These additives either alone or in combination with others resolve the complex secondary structure formation at GC-rich regions (Henke et al., 1997; Musso et al., 2006; Ralser et al., 2006), or they enhanced the amplification possibilities of GC-rich templates. It was also shown that template denaturation with NaOH, hot start PCR, step-down PCR, and primer modification can improve PCR amplification of highly GC-rich regions of DNA (Agarwal and Perl, 1993; Sahdev et al., 2007). These efforts to amplify GC-rich regions are found useful in some but not in all the cases (Mamedov et al., 2008) and highlight the need to standardize GC-rich gene specific protocol to overcome the problem.
Protamine (PRM) is one of the genes having very high GC content (88% in the coding region) in chicken (Oliva and Dixon, 1989). The chicken PRM gene is intron less (Oliva and Dixon, 1989), strikingly similar to mammalian protamines (Nakano et al., 1976). Most recently, PRM transcripts in sperm has emerged as fertility/infertility biomarkers (Miller, 2011; Shafeeque et al., 2014a). However, their role in avian sperm is not studied in detail (Shafeeque et al., 2014b), most probably because of difficulties in amplifying avian protamine gene due to its high GC content. Therefore, the aim of this study was to develop an easy PCR protocol to amplify the GC rich PRM.
MATERIALS AND METHODS
The protocols involving the care and use of animals for these experiments were in accordance with the rules of the ‘Animal Ethics Monitoring Committee of the Central Avian Research Institute’.
Experimental Birds
In the current study, adult male (35 weeks old) broiler breeders (IC 3) from the same hatch were used. The birds were housed in individual cages and were maintained under uniform standard management conditions with 14 h light: 10 h dark.
Sample Collection
Four birds were sacrificed and a small piece (approx 1cm long) of testes from each bird was collected aseptically and washed in chilled RNase free PBS. Special care was taken to remove blood from the samples. Each collected sample was divided into 2 parts: the first part was used immediately for RNA isolation while the second part was immersed in RNAlater and stored at –4°C.
RNA Isolation and Reverse Transcription
Total RNA from individual sample was extracted by RNAzol® RT (Molecular Research Centre, Inc., Cincinnati, USA) according to the manufacturer’s instructions. Approximately 25mg tissue samples were used for RNA isolation. An optional phase separation step by using 4-bromoanisole was performed during total RNA isolation to eliminate gDNA contamination. The concentrations and purities of isolated RNA samples were determined spectrophotometrically at A260 and A280nm. For all samples, the RNA 260/280nm absorbance ratio was ≥2.0, and concentration was approx 4500ng/µl. Further, all the RNA samples were used for PCR with β-actin primer to check DNA contamination. DNA free total RNA samples (1µg) were reverse transcribed using the RevertAid First Strand cDNA Synthesis Kit (MBI Fermentas, Hanover, MD, USA) according to the manufacturer’s instructions. The resultant cDNA was stored frozen at –20°C until used.
Protamine Amplification by PCR
Exonic region of PRM gene (187 bp) was amplified using a pair of gene specific primer (F: 5′-CGCAGCAGGACCCGCAGCCG-3′ and R: 5′-CGGCGGCGGCGGCTCAGTAG-3′) with available three different Taq DNA Polymerases (Go Taq from Promega, USA; Taq DNA polymerase from Geneaid, Taiwan; and hot start Taq DNA polymerase from Fermentas, Canada). The polymerase chain reaction (PCR) was carried out in a total volume of 25 μl containing 4μl of 10 times diluted first strand of cDNA, 1X PCR buffer (Geneaid, Taiwan), 1.5mM MgCl2 (2.0 mM for Geneaid Taq), 2.5 mM of dNTPs, 5 pM of each primer and 0.625 unit of Taq DNA polymerase for Go Taq and Hot Start Taq (one unit for Geneaid Taq) in presence and absence of 8% DMSO. Gradient PCR methodology was used to ascertain the optimum annealing temperature of protamine primer pair for successful amplification of PRM gene. After the initial denaturation at 96oC for 5 min, the PCR profile consisted of a denaturation step at 97oC for 20sec, an annealing step from 60.6 to 67.5oC for 30sec with and an elongation step at 74oC for 20sec in total for of 34 cycles, followed by a final extension of 5min at 74oC. The PCR product was run on 1.5 % agarose gel including EtBr in 1X TAE buffer. The gel was visualized under UV light and was photographed using automated gel documentation system. The concentration of buffer, MgCl2, DMSO, template and hot / normal start were optimised for the Go Taq DNA Polymerase.
RESULTS
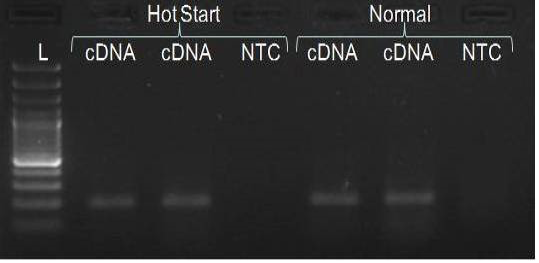
We have amplified PRM amplicon successfully at all the given temperatures (60.6-67.5) using 1.5mM MgCl2 and Go Taq DNA polymerase only in presence of 8% DMSO (Figure 1.1). However, it could not be amplified productively at any given temperature by using hot-start Taq DNA Polymerase from Fermentas (Figure 1.2) or Taq from Geneaid (Figure 1.3) in presence /absence of DMSO. To find out optimal buffer concentration, separate PCR reactions were setup with addition of 1X, 0.8X, 0.6X, 0.4X, 0.2X buffer and without buffer. Results indicated that 0.8X buffer (Figure 2) concentration was optimum for good amplification. Further, concentration of MgCl2 was optimized in separate PCR reactions with addition of 1.0mM, 1.4mM, 1.8mM, 2.2mM, 2.6mM and 3.0mM MgCl2. Good amplification was achieved at 1.4mM MgCl2 (Figure 3). Additionally DMSO concentration was optimized at optimum buffer and MgCl2 concentration in a separate PCR reaction with addition of 4.0%, 6%, 8%, 10%, 12% and 14% DMSO. Final concentration of 10% DMSO was the only one to provide the desired amplicon yield without nonspecific amplification (Figure 4). Furthermore, concentration of template (1-6µl of 10 time’s diluted cDNA) was also optimized in a new PCR reaction with optimum concentrations of buffer, MgCl2 and DMSO. The optimum template concentration was 5µl (Figure 5) to obtain best PCR efficiency. Finally, effect of hot start and normal start PCR was observed by two separate reactions, one kept at cold PCR block while running the PCR and another kept directly at Hot (96°C) block while running the PCR. Results indicated that product amplified using Hotplate has very less primer dimer (Figure 6).
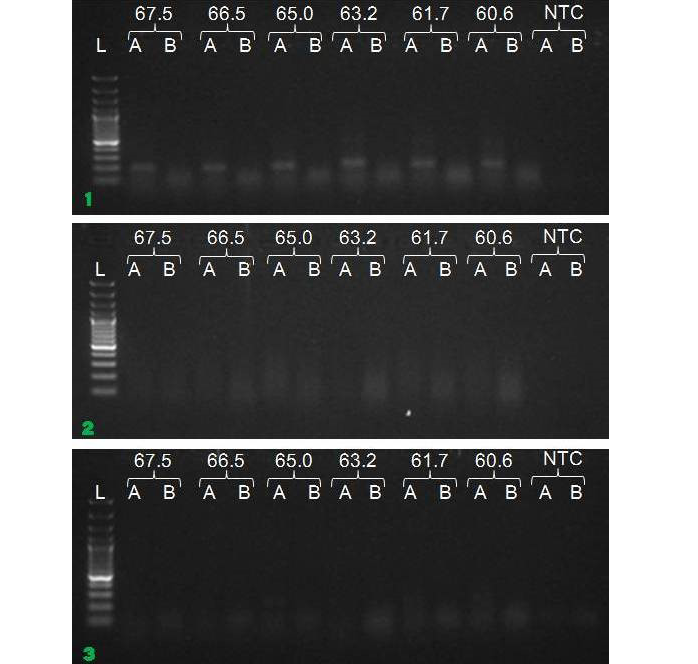
Figure 1: Amplification of PRM with Go Taq (1.1), Hot start Taq (1.2) and gene aid (1.3) (L=100 bp DNA Ladder, A=With DMSO, B= Without DMSO)
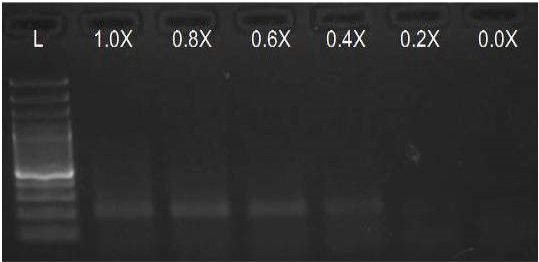
Figure 2: Amplification of PRM with Go Taq using different buffer concentrations (L= 100 bp DNA Ladder)
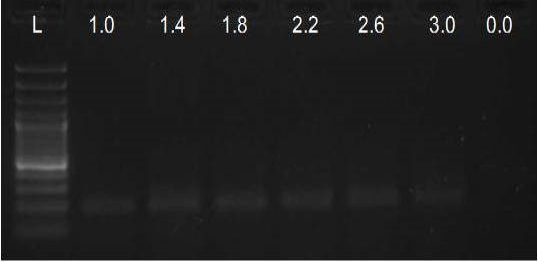
Figure 3: Amplification of PRM with Go Taq using different MgCl2 concentrations (L= 100 bp DNA Ladder)

Figure 4: Amplification of PRM with Go Taq using different DMSO concentrations (L= 100 bp DNA Ladder)

Figure 5: Amplification of PRM with Go Taq using different template concentrations (L= 100 bp DNA Ladder)
DISCUSSION
Determining the most suitable direct PCR DNA polymerase for detecting a high GC rich target is time-consuming and costly. However, our data highlight the importance of comparing the performance of three DNA polymerases prior to establishing PCR conditions. The results indicated that among the commercially available polymerases we tested; only Go Taq DNA polymerase has the potential for the amplification of more than 80 percentage GC rich region. We optimised different PCR conditions for Go Taq DNA polymerase, which are as follows.
To optimize the PCR thermal cycling conditions, one has to determine the optimal temperature and length of each of the program segments, as well as the number of cycles. Of those, the most important parameter seems to be the temperature of primer annealing, as even the smallest deviation of 1°C or 2°C could make a significant difference between specific and nonspecific amplification (Roux, 2009; Lo and Chan, 2006). The optimal primer annealing temperature for a particular PCR amplification depends on the base composition, nucleotide sequence, length and concentration of the primers (Saiki, 1989). A typical primer annealing temperature is 5°C below the calculated Tm of the primers. Increasing the annealing temperature enhances discrimination against incorrectly annealed primers and reduces mis-extension of incorrect nucleotides at the 3′ end of primers. Therefore, a higher annealing temperature increases amplification specificity. In general, the annealing temperatures usually range between 55°C and 72°C. However, since the G–C pair is bound by three hydrogen bonds, while A–T pairs is bound by only two, high GC content corresponds to higher melting temperature and requires higher temperature for primer annealing (Kramer and Coen 2001; Mamedov et al., 2008). In the present study, the annealing temperature was determined to 67.5°C in presence of DMSO. Gradient PCR reaction showed that the optimal annealing temperature for our PCR reaction was between 60.6°C to 67.5°C.
We optimised the strength of the PCR buffer supplied with the Taq DNA polymerase and found that 0.8 buffer strength was good for best amplification of PRM amplicon instead of the recommended buffer strength. Reason behind this may be that the melting temperature of DNA increases with higher ionic strength.
In the present study, the MgCl2 concentration tested was within the range of 1.0 to 3.0mM, and the optimum observed was 1.4mM. Yet, up to 2.2mM MgCl2 also resulted in satisfactory amplification, which gave best results when performed on PCR products obtained with 1.4mM MgCl2 concentration. The observed range of acceptable molarities was broad most probably due to the presence of enhancer DMSO, known to improve the success of PCR reaction even at different MgCl2 concentrations (Kramer and Coen, 2001).
Addition of DMSO to the reaction mixture turned out to be essential for successful amplification, and none of the previously described optimization strategies could have been implemented without its presence. Optimal concentration of 10% was determined after testing three different options, which was within the recommended range of 1–10% (Grunenwald, 2003; Kolmodin and Birch, 2002).
The quality and concentration of DNA templates can directly affect the outcome of PCR amplifications. To achieve satisfactory amplification, certain baseline conditions may be used as a starting point for optimizing PCR amplification. For a typical PCR, 104 to 107 molecules/100 to 500ng of template DNA is recommended. There are several methods for purifying DNA for PCR amplification, including commercially available kits, as well as standard methods (Sambrook et al., 1989). Analysis by pulsed-field agarose gel electrophoresis is typically recommended for template DNA used in long PCR to assure its purity and integrity.
In the present study, all PCR components was mixed at a lower temperature on ice and then PCR block was heated to 96°C; PCR reaction tubes transferred directly to hot block from ice. Adopting this procedure we observed a significant decline in primer dimer formation in comparison to PCR run as usual.
CONCLUSION
PRM amplicon successfully amplified only with Go Taq DNA polymerase in presence of DMSO. The optimization of the PCR conditions included determination of optimal MgCl2 concentration, buffer strength and annealing temperature, in presence of 10% DMSO and the DNA template of acceptable concentration, resulted in successful amplification. In addition, PCR started directly at hot plate is also important to get rid of primer-dimer formation.
ACKNOWLEDGEMENT
The authors gratefully acknowledge the Science and Engineering Research Board (SERB), Department of Science and Technology, Government of India for supporting this research program (SERB/FT/LS–147/2011).
REFERENCES

