

A Homozygous c.1131G>A Missense Mutation in BBS9 Gene Manifesting Autosomal Recessive Bardet-Biedl Syndrome in Consanguineous Kashmiri Family




A Homozygous c.1131G>A Missense Mutation in BBS9 Gene Manifesting Autosomal Recessive Bardet-Biedl Syndrome in Consanguineous Kashmiri Family
Syeda Ain-ul-Batool1, Sadia1, Kathrin Blasius2,3,4, Angela Kaindl2,3,4 and Ghazanfar Ali1,*
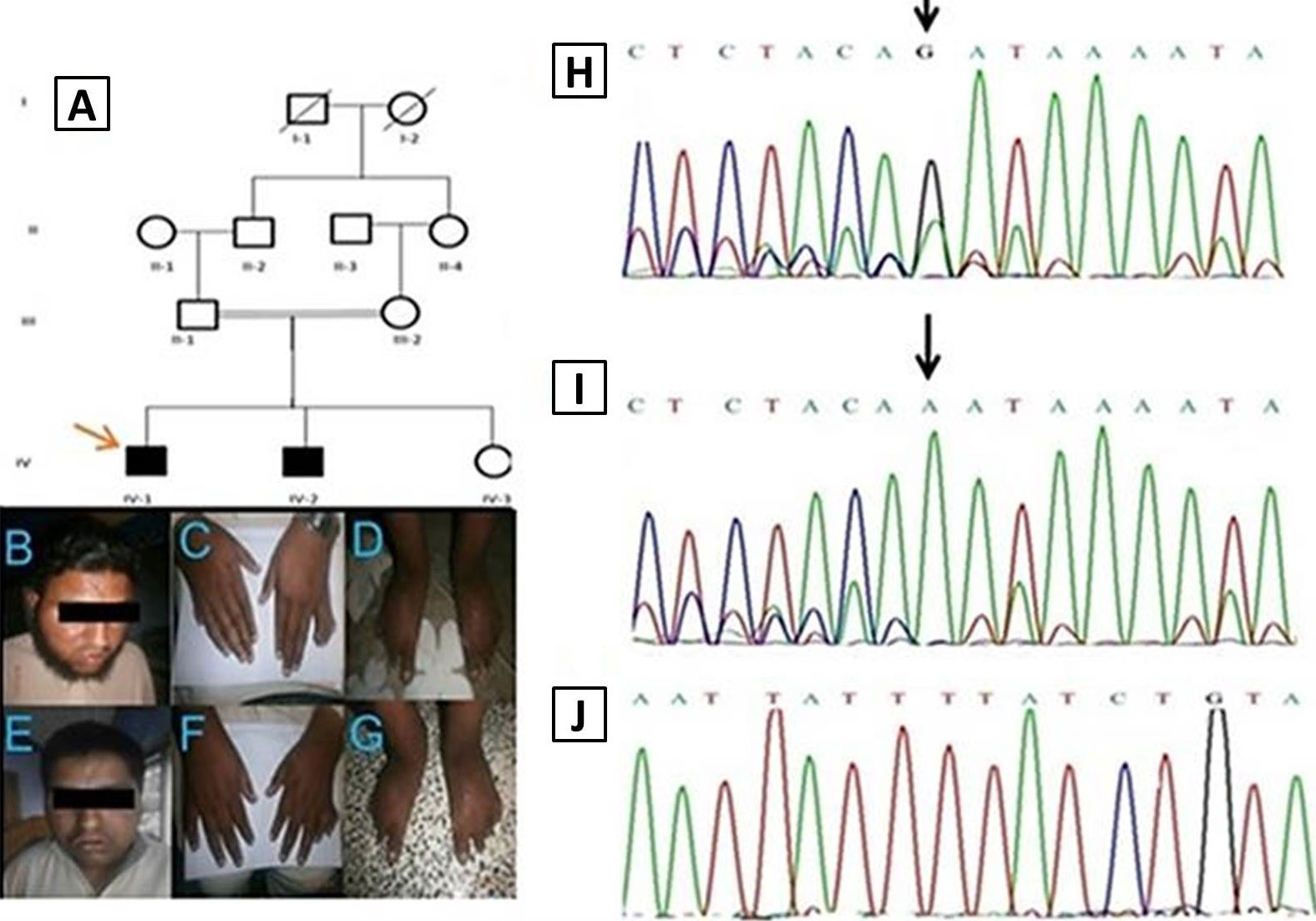
A, pedigree of family (MR20) showing autosomal recessive inheritance, while red arrow indicates the affected individual for whom WES was done. B, affected individual IV-1, having typical features of BBS syndrome including hypertelorism, eyes exotropia and a flat nasal bridge. C, dorsal view of hands showing polydactyly in right hand while polydactyly and syndactyly is clearly visible in left hand. D, dorsal view of feet having polydactyly in both. E, typical BBS facial features shown by affected individual IV-2, having flat nasal bridge, poor eye sight, intellectual disability and small mouth. F, dorsal and palmar view of hands, and having post axial polydactyly in left hand. G, feet of affected individual IV-4, showing obesity and polydactyly in left foot. H, homozygous mutation in the patient IV-1 showing splice acceptor site change G>A on position 167. I, heterozygygous carrier and J, homozygous Normal.




{kind=link}