Gut Microbe Analysis between Asthma Patients and Healthy Volunteers in Shaanxi Province, Xian, China





Gut Microbe Analysis between Asthma Patients and Healthy Volunteers in Shaanxi Province, Xi’an, China
Hafiz Muhammad Ishaq1, Muhammad Shahzad2, Xiaokang Wu3, Chaofeng Ma4 and Jiru Xu1,*
1Department of Microbiology and Immunology, Key Laboratory of Environment and Genes Related to Diseases of Chinese Ministry of Education, School of Medicine, Xian Jiaotong University, Yanta West Road No. 76, 710061 Xi’an, Shaanxi Province, China.
2Department of Pharmacology, University of Health Sciences, Khyaban-e-Jamia Punjab, Lahore
3The Second Affiliated Hospital of Xi’an Jiaotong University, 157 Xiwu Street, Xi’an China
4Xi’an Center for Disease Control and Prevention, China
ABSTRACT
Asthma is an allergic and chronic lung disease that causes inflammation and narrows the air passages. Present study aimed to characterize the gut microbiota of asthma patients and to investigate the alteration in similarity and diversity of gut microbial composition with comparison of healthy controls. Fecal samples from fifteen patients and five healthy individuals were collected. PCR-DGGE, by using universal primers focusing V3 region of 16S rRNA gene, was performed for characterization of gut microbial composition. Sequencing of most dominant excised gel bands was done. Real-time PCR was performed to evaluate the copy numbers of dominant bacteria of gut microbiota. The results indicated that a significant diversity difference between asthma and healthy groups (*P< 0.05), was shown in DGGE profile. Similarity index was found to be lower in inter-group than intra-group that indicated a change in composition of gut flora in asthma patients. Sequencing results also indicated a change of bacterial composition between both groups. qPCR results showed a significant decrease in Bifidobacterium, Lactobacillus and Clostridium leptum sub group (*P<0.05) and non-significant decrease and increase in Bacteroides vulgatus and Peptostreptococcus productus, respectively. Summarizing all, in asthma patients, there is a significant change in the molecular characterization of gut microbiota.
Article Information
Received 26 October 2016
Revised 03 April 2017
Accepted 08 June 2017
Available online 04 January 2018
Authors’ Contribution
HMI performed all the experiment and prepared initial draft of the manuscript. MS revised the manuscript, XW guided to collect the samples for this study and CM helped to collect the samples. JX supervised the research project and provide guidance and support to do research.
Key words
Asthma, Gut microbiota, Real Time PCR, DGGE profile.
DOI: http://dx.doi.org/10.17582/journal.pjz/2018.50.1.165.173
* Corresponding author: xujiru@mail.xjtu.edu.cn
0030-9923/2018/0001-0165 $ 9.00/0
Copyright 2018 Zoological Society of Pakistan
Introduction
Human gut microbiota is an important factor in determining the health status of the host. Its composition and functional activities are stable over time but may be altered by different factors including age, disease and diet (Power et al., 2013). Human gut flora contains approximately 100 trillion bacteria and plays a vital role in human physiology including metabolism, nutrition absorption and immune function (Walsh et al., 2014; Ishaq et al., 2017a). Gut microbiota plays an important role in host immune homeostasis and produces defense against pathogens (Kamada et al., 2013; Tan et al., 2013).
Modulation of gut microbial composition has been associated with many other diseases i.e. colitis, Crohn’s disease, hyperthyroidism, viral diarrhea, inflammatory bowel disease, metabolic diseases such as obesity, and type II diabetes (Ma et al., 2011; Zhou et al., 2014; Khoruts et al., 2010; Ishaq et al., 2017b).
Asthma is an allergic chronic inflammatory disease that causes a substantial burden on patients and their families (Bonamichi-Santos et al., 2015). Over the last decade, asthma prevalence has dramatically increased in Western societies. It is a complex disease having strong genetic and environmental components that manifests with a variety of clinical features i.e. short breathing, cough, chest congestion, wheezing and sputum production (Huang and Boushey, 2015). A dysbiotic gut flora may have an important role for developing allergic diseases (Muir et al., 2016).
Denaturing gradient gel electropheresis (DGGE) of PCR amplified 16S rRNA genes has been performed to evaluate the gut microbial diversity (Wang et al., 2016). It gives a DNA fingerprinting of experimental samples; also provides the help to identify the bacterial community through sequence analysis and to check their dynamics. For the characterization of complex bacterial communities, qPCR and 16S rRNA gene libraries are marvelous development (Dorigo et al., 2005).
PCR–DGGE along with image analysis helps to study the microbial similarity and diversity, while erection of dendrogram (UPGMA) and analysis of sequence were done to study the disease linked DGGE motifs and taxa (Wu et al., 2010). Real-time PCR was performed by using primers of Clostridium leptum sub group, Bacteroides vulgatus, Peptostreptococcus productus, Lactobacillus and Bifidobacterium genera for quantitative evaluation of gut microbiota of asthma patients.
The aim of current study was the characterization of gut microbiota of asthma patients and to investigate and study the alteration in similarity and diversity of gut microbial composition in comparison with healthy controls.
Material and methods
Sample collection and ethics statement
A detailed semi-structured questionnaire was given to each volunteer after they had received and read a complete description of the study. All participants provided written informed consent and all the protocols were approved by the Ethics Committee of School of Medicine, Xi’an Jiaotong University. Additionally, the research and recruitment protocols were carried out according to the Ethical Principles for Medical Research Involving Human Subjects adopted in the Declaration of Helsinki by the World Medical Association. All samples used in the study followed standardized rules governing sample handling, and information obtained in the interview was recorded on standardized data collection forms.
Fecal samples were collected from 15 asthma (7 female and 8 males, having age between 30 to 45 years) and 5 healthy volunteers (2 females and 3 males, having same age between 30 to 45 years) in a sterile cup. A questionnaire was filled regarding age, gender, dietary habits, body weight, and health status of patients as well as healthy volunteers. Two patients were tracked for about three weeks to check the stability of gut microbiota (samples collected on day 1, 7, 14 and 21days). All the samples were delivered on ice, usually within 4 hour of defecation. Upon arrival in the laboratory, fecal sample were stored at -80 ºC until DNA extraction. None of the patients and healthy individuals had any record of gastrointestinal disease; also, there was no history of using prebiotics, probiotics and antibiotics, two months prior to sampling.
DNA extraction from fecal sample
After thawing of all fecal samples, DNA was extracted with the help of QIAGEN QIAamp Stool kit (QIAGEN, Germany) as per instructions of manufacturer, with initial start of bead-beating step at 5000 rpm for 30 sec. Concentration of DNA was evaluated by the use of NanoPhotometer TM (IMPLEN, Germany) (Scanlan et al., 2006).
PCR amplification for DGGE
Fecal bacterial DNA was used for PCR–DGGE. Universal primers (Table I) were used to amplify the 16S rRNA gene (V3 region). 50 μl PCR reaction mixture contain 20 pmol of two primers, 2.5 mM 10×buffer, MgCl2 2.5 mM, Taq (Promega, USA) DNA polymerase 2 U, deoxy nucleotide triphosphate (dNTP) 200 mM and 120 ng DNA approximately 2 μl. An automatic thermal cycler (ABI2720 made in USA) was used to amplify the PCR mixture through PCR touchdown programming: initial start with denaturation of PCR mixture at 95 ºC for 5 min, then denature by 95 ºC for 1 min, annealing temperature reduced to 65 ºC for 1 min and then to increase 72 ºC for 1 min. Decrease of annealing temperature 1ºC in every second cycle so that touchdown temperature become 55 ºC, at this stage of temperature, 10 extra cycles were carried out, and then followed by 72 ºC for 7 min for final extension and finally maintained at 4 ºC. 1.5% agarose gel were used to electrophoresis the amplified PCR mixture and dipped in ethidium bromide solution to visualize under UV light (Muyzer et al., 1993).
Table I.- Primers used in PCR-DGGE.
| Primer | Sequence (5¹–3¹) |
| 341-F | CCTACGGGAGGCAGCAG |
| 534-R | ATT ACCGCGGCTGCTGG |
| 341FG |
CGCCCGCCGCGCGCGGCGGC GCGGGGCGGGGGCACGGG GGGCCTACGGGAGG CAG CAG |
Reference: Matsuki et al. (2004).
Denaturing gradient gel electrophoresis
In brief, amplified PCR product of total bacteria was loaded in 8% (w/v), acrylamide-bis, 37.5:1, gels in a 1×TAE buffer solution tank, having 30~65 % linear denaturant gradient. The gel was run for 13 h at 90 V with constant temperature at 60ºC. Calculation of bacterial diversity of each sample was done by the total number and the strength of DGGE bands, with the help of Quantity one software. A similarity index was studied through Dice’s similarity coefficient (van Der Gucht et al., 2001). UPGMA (arithmetic averages) through unweighted pair group method, was used to establish a dendrogram (Ling et al., 2010).
Statistical analysis of DGGE band pattern
The number of bands and band intensity of DGGE profiles was determined by using Quantity one software. Taxonomic diversity of bacteria was evaluated by Shannon–Weaver index of diversity (H¹) (Gafan et al., 2005; Ledder et al., 2007). Similarity index and DGGE profile’s cluster analysis were done through UPGMA method (band based Dice similarity coefficient). As there was no uniformity in data distribution, Mann–Whitney U test was used for a nonparametric statistical analysis, where (P<0.05) was considered as statistically significant. Arithmetic averages (UPGMA) and clustering algorithm were used to calculate the dendrograms for unweighted pair group method (Fromin et al., 2002; Silva and Russo, 2000). To observe the stability of gut microbial composition, two patients were checked for a period of 3 weeks; gut microbial DGGE band analysis showed high similarity in bands patterns. Dice similarity coefficient ranged between 96-97% (Figure not shown).
Calculation of Shannon–Weaver index (H¹) was done with the help of following equation:
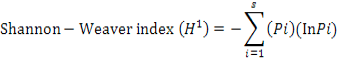
Excision of bands and sequencing
Physically a sterilized scalpel was used to excise the dominant band of interest from the gel with care. The polyacrylamide gel piece was placed in a tube containing 50 µl of water and incubated at 37 ºC for 30 min. After centrifugation, 8 μl of DNA template was used to re-amplify the V3 region of 16S rRNA gene by using original primers (without GC clamp) as previously used for DGGE analysis (Green et al., 2010). Sequencing of re- amplified PCR products were done by ABI 3500xL. Obtained Sequences were analyzed by using BLAST software for identification of species or genus (McBain et al., 2003).
Real-time PCR
Real time PCR quantification was performed in a CFX96 Bio-Rad (USA). Total 20 μl reaction mixture contained 1 μl of each primer (5 uM), 10 μl of 2× SYBR Green (TOYOBO, Japan), 2 μl sample DNA and 6 μl sterilized H2O. Amplification program composed of 1 cycle at 95 ºC for 5 min, then followed by 40 cycles at 95 ºC for 10 sec. then 55 ºC for 15 sec. and followed by 72 ºC for 50 sec. and finally 1 cycle at 95 ºC for 15 sec. The fluorescent signals were detected at the end of every cycle. After amplification, PCR melting temperature analysis was done to evaluate the specificity of the PCR system. Through slow heating at 0.1 ºC/s with increase from 65 ºC to 95 ºC, melting curves were achieved, with continuous fluorescence deduction. Real time PCR Primers were shown in Table II. To determine the copy number of Clostridium leptum sub group, Peptostreptococcus productus, Bacteroides vulgatus, Lactobacillus and Bifidobacterium genus in each DNA samples, in same experiment, fluorescent signals were calculated, averaged and compared with standard curve that were generated with standard DNA from six serial dilutions in linear range of the program (Ma et al., 2011; Wu et al., 2010). Bacteroides vulgatus (CICC 22938), Bifidobacteriaum (CICC 6186), NWS Lactobacillus, Peptostreptococcus productus (from our lab) and Clostridium leptum sub group (YIT 6169) were used as standard strains. Real time PCR was done thrice and mean was considered in results. Data were reported as the average estimate of logarithms of fecal PCR target genetic amplicon copy numbers present in 1g of feces.
Statistical analysis
Real time PCR and PCR-DGGE experiments were repeated thrice. Statistics software SPSS 12 was applied for statistical analysis. P values were calculated by using the U (Mann-Whitney) test. Results were considered as statistically significant, where (P<0.05).
Table II.- Primers that were used in real time PCR.
| Target bacteria |
Primer Sequence (5¹–3¹) |
Band size | |
| Bifidobacterium | Bifid F | CTC CTGGAAACGGGTGG | 550 bp |
| Bifi-R | GGTGTTCTTCCCGATATCTACA | ||
| Lactobacillus | LactF | CTC AAA ACT AAACAAAGTTTC | 250 bp |
| LactR | CTC AAA ACT AAACAAAGTTTC | ||
| Bacteroides vulgatus | BV- F | GCATCATGAGTCCGCATGTTC | 287 bp |
| BV-R | TCC ATA CCC GACTTT ATT CCTT | ||
| Clostridium leptum | C.lep-F | GCACAAGCAGTG GAG T | 239 bp |
| C.lep-R | CTTCCTCCGTTTTGTCAA | ||
| Peptostreptococcus productus | PSP-F | AACTCCGGTGGTATC AGA TG |
268 bp |
| PSP-F |
GGGGCTTCT GAG TCAGGTA |
Reference: Matsuki et al. (2002), Dubernet et al. (2002) and Wang et al. (1996).
Results
DGGE profiles of asthma and healthy group
DGGE analysis was done with amplified PCR product by using universal primers targeting the 16S rRNA (V3 region) gene in both healthy and asthma group. In Figure 1, D1–D15 indicate samples from asthma and C1-C5 healthy controls. As the position, number, and bands intensity were different among experimental samples, which showed the complex intestinal microbial fingerprints. In DGGE gel, there were some common bands and some were less intense bands present in different samples, at the same position but in different lane. A total 74 bands were detected by using Quantity one software (van Der Gucht et al., 2001), in 15 tracks of asthma with average band of (5.7 ± 2.015). A total 34 bands were detected in 5 tracks of control group with average of (6.8 ± 1.095). For analysis of gut microbial composition in asthma and healthy group, U (Mann-Whitney) test was applied for comparison of Shannon weaver index (H¹) of diversity. The diversity (H¹) results indicated (2.00 ± 0.31 vs. 2.2 7 ± 0.21) significant (P < 0.046*) difference between the two groups. Similarity index results showed to be lower in inter-group than intra-group that indicated a change in composition of gut flora in asthma patients. DGGE profiles similarity was determined through Dice similarity coefficient and dendrogram UPGMA shown in Figure 2. The band-based Dice similarity coefficient values of asthma and healthy group and with the mean similarity index were (0.235 ± 0.200) and (0.219 ± 0.151) respectively, shown in Table III. When all the values of statistical samples of asthma and the healthy group were compared by Dice similarity coefficient, mean similarity index was (0.205 ± 0.127) between two groups, which showed that it was to be lower in inter-group than intra-group, which shows that gut microbial composition of asthma patients was different from healthy control group.
Sequencing results analysis of dominant band
In Figure 1, 16 bands from DGGE gel were excised for quantity analysis. To certify the resolution capability of DGGE bands in different lane but same positions, the bands C4a and D13a, D10a, D11a and D12a were cut for sequence analysis. Bands C4a and D13a were identified as Bacteroides vulgatus with 95 % similarity while, bands D10a, D11a and D12a were identified as Klebsiella oxytoca with 94 % similarity. Taxonomic identity of other bands has been shown in Table IV. Sequencing results were analyzed by using BLAST. Results indicated that phyla Firmicutes, Bacteroidetes, and Proteobacteria were dominant.
Real time PCR quantification
The Bifidobacterium, Peptostreptococcus productus, Bacteroides vulgatus, Clostridium leptum and Lactobacillus were quantified by real time PCR. Results showed that the copy number of Bifidobacterium (6.10 ± 0.25 vs. 6.54 ± 0.67), Lactobacillus (7.61 ± 1.05 vs. 8.65 ± 0.09) and Clostridium leptum sub group (4.32 ± 0.28 vs.
Table III.- Gut microbial similarity and diversity of asthma patients and healthy subjects.
| Groups |
Diversity |
|
Similarity |
||
|
Number of bandsa |
Shannon indexb |
Intra-similarityc |
Inter-similarityd |
||
| Disease group |
5.7 ± 2.015 |
2.00 ± 0.31 |
|
0.235 ± 0.200 |
0.205 ± 0.127 |
| Control group |
6.8 ± 1.095 |
2.27 ± 0.21 |
|
0.219 ± 0.151 |
|
| P Value |
0.132 |
0.046* |
|
- |
- |
Results that are significantly different with P<0.05 (Mann–Whitney U test); aBands numbers, denaturing gel electrophoresis (DGGE) produced by each sample; bShannon diversity index (H¹) was calculated by using the relative intensities of all DGGE bands present in each sample; cComparing of DGGE band profiles within individual of a given groups by Dice similarity coefficients; dComparing of DGGE band profiles between members of asthma and healthy groups by Dice similarity coefficients.
Table IV.- Sequences of excised PCR Amplicons from DGGE gel and identities based by BLAST database.
|
Selected excised bands |
Bacteria with highest % homology |
Sequence accession number |
Bacterial phyla | Gene bank number |
| D2a | Sporomusa ovate (89) | DSM 2662 | Fermicutis | NZ_ASXP01000005.1 |
| D3a | Escherichia coli (98) | MG1655 | Protobacteria | NC_000913.3 |
| D3b | Bacteroides uniformis (98) | CL03T00C23 | Bacteroidetes | NZ_JH724260.1 |
| D5a | Alistipes putredinis (89) | DSM 17216 | Bacteroidetes | NZ_DS499580.1 |
| D5b | Ruminococcus flavefaciens (87) | MC2020 | Fermicutis | NZ_JNKE01000007.1 |
| D6a | Prevotella copri (95) | CB7 | Bacteroidetes | NR.040877.1 |
| D7a | Bacillus pumilus (87) | NJ-M2 | Fermicutis | NZ_CP012329.1 |
| D7b | Bacteroides pyogenes (90) | JCM 10003 | Bacteroidetes | NZ_BAIU01000058.1 |
| D10a | Klebsiella oxytoca (94) | CAV1374 | Protobacteria | NZ_CP011636.1 |
| D11a | Klebsiella oxytoca (94) | CAV1374 | Protobacteria | NZ_CP011636.1 |
| D12a | Klebsiella oxytoca (94) | CAV1374 | Protobacteria | NZ_CP011636.1 |
| D13a | Bacteroides vulgatus (95) | ATCC 8482 | Bacteroidetes | NC_009614.1 |
| D14a | Parabacteroides distasonis (90) | ATCC 8503 | Bacteroidetes | NZ_BAIU01000058.1 |
| C1a | Prevotella histicola (90) | F0411 | Bacteroidetes | NZ_JH376764.1 |
| C4a | Bacteroides vulgatus (95) | ATCC 8482 | Bacteroidetes | NC_009614.1 |
| C5b | Bacteroides coprocola (97) | DSM 17136 | Bacteroidetes |
NZ_DS981502.1 |
Table V.- Real time PCR quantification (Mean ± SD) of different bacteria.
| Bacteria |
Healthy subjects |
Patients |
P value |
|
Bifidobacterium (105) |
6.54±0.67 |
6.10±0.25 |
0.020* |
|
Bacteroides vulgatus(108) |
2.30±1.51 |
1.90±1.05 |
0.319 |
|
Lactobacillus (106) |
8.65±0.09 |
7.61±1.05 |
0.021* |
|
Clostridium leptum(106) |
4.59±0.19 |
4.32±0.28 |
0.031* |
|
Peptostreptococcus productus (102) |
1.40±1.00 |
1.80±0.90 |
0.334 |
Results were reported through the average estimate of logarithms in fecal PCR target genetic amplicon, copy numbers present in 1 g of feces* indicates (P<0.05).
4.59 ± 0.19) were significantly (*P<0.05) decreased in the patients as compared to healthy subjects. While the copy number of Peptostreptococcus productus (1.80 ± 0.90 vs. 1.40 ± 1.00) and Bacteroides vulgatus (1.90 ± 1.05 vs. 2.30 ± 1.51) were non-significantly (P<0.05) increased, and decreased respectively, in the fecal samples of patients as compared to healthy subjects. All these results are summarized in Table V.
Discussion
Human gut microbiota plays a vital role to protect the body against diseases by performing metabolic, trophic and protective function (Guarner and Malagelada, 2003). Gut microbial composition can be altered in many diseases like Crohn’s disease, malnutrition, inflammatory bowel disease, colitis, hyperthyroidism, obesity and type II diabetes (Ma et al., 2011; Zhou et al., 2014; Khoruts et al., 2010). Our study showed that there was difference between gut microbial composition of asthma patients and healthy subjects as determined by sequence of dominant bands from DGGE profile and real time PCR analysis. More precisely, there were significant differences in the diversity and similarity of bacterial communities, measured by Shannon-weaver index (H¹) and Dice similarity coefficient. The bacterial copy number of Bifidobacterium, Lactobacillus and Clostridium leptum sub group were significantly decreased in asthma patients group as compared to control group. While Peptostreptococcus productus and Bacteroides vulgatus were non-significantly increased, and decreased in diseased group as compare to healthy controls, respectively. In current study, in asthma patients group and healthy control group, bacterial similarity and diversity of the gut microbiota were analyzed by combining DGGE profile (targeting 16S rRNA gene) with imaging and sequencing of dominant PCR amplicons, together with statistical analyses. DGGE fingerprint techniques focusing 16S rRNA gene and quantification of major bacteria with real time PCR have been used to study the complex bacterial communities (Zhou et al., 2014; Ma et al., 2011). To evaluate the diversity of gut microbial composition, in asthma and control group, we calculated the Shannon–Weaver index (H¹). Compared to healthy individuals, (H¹) was significantly lowered in asthma group (P < 0.046*). The decreased bacterial diversity might have altered the gut microbial composition in asthma. While similarity index was found to be lower in inter-group than intra-group, which showed that gut microbial composition was changed in asthma patients group. In our study, gut microbial diversity, Shannon–Weaver index (H¹), results are consistent with previous study of gut microbe analysis of hyperthyroid patients (Zhou et al., 2014). It shows that gut microbial composition of patients varies to different degree as a result of asthma occurrence. Hence, asthma may seriously influence the composition of gut microbiota. These results indicate that due to occurrence of asthma, it may cause the physiological intestinal changes which may result in alteration of gut microbial composition. Furthermore, the change in gut microbial composition may lead to the worsening of sickness (Kamada et al., 2013).
Previous studies on type II diabetic patients, hyperthyroidism and obesity, found the dominance of phyla Bacteroidetes, Firmicutes and Protobacteria in gut microbiota (Wu et al., 2010; Gill et al., 2006; Ley et al., 2005; Zhou et al., 2014). Our results also showed the same phyla Bacteroidetes Firmicutes and Protobacteria in the gut microbiota of asthma patients. According to sequence results, Klebsiella oxytoca, found in asthma patients group, which is a gram-negative bacterium that can cause colitis and sepsis (Högenauer et al., 2006).
DGGE has many potential advantages over culture techniques for anaerobic gut microbial analysis. Multiple samples can be analyzed, simultaneously, on a single gel which gives a direct comparison among samples, and dominant bands of interest can be excised and sequenced directly to identify bacterial species or genus. To analyze the molecular characteristics of complex microbial communities, DGGE fingerprint has, now, routinely been used (Ma et al., 2011; Wu et al., 2010; Zhou et al., 2014). Though, DGGE is a semi-quantitative technique, band density results may not accurately relate to the target abundance, hence, there is possibility that a clear associations between species abundance and diseases presence may not necessarily be recognized (Ledder et al., 2007). In our study, some significant and basic gut microbial characterization were established by combining similarity and diversity analysis with dominant DGGE bands excision and with PCR re-amplification of excised bands and sequencing. However, there was a significant disparity in intestinal microbial composition between asthma patients and normal control, established in the current study was fairly interesting as there was no direct relationship between asthma and intestinal flora. So, present study findings of dissimilar gut bacterial community texture between asthma and control groups shows that alteration of health condition could influence the gut microbial composition.
We also performed real time PCR to observe the accurate quantitative changes of intestinal microbiota (Lyons et al., 2000) and results indicated that, there was significant reduction of Bifidobacterium and Lactobacillus in diseased group as compared to healthy control which are aligned with previous work of patients with hyperthyroidism in which Bifidobacterium and Lactobacillus significantly reduced (Zhou et al., 2014). Also, there was a significant decrease of Clostridium leptum sub group in asthma patients which is aligned with previous study of Clostridium leptum sub group bacteria abundance and diversity in stool microbiota of inflammatory bowel disease patients, furthermore, this shows low level of Clostridium leptum sub group causes inflammatory bowel disease (Kabeerdoss et al., 2013). Moreover, there was non-significant increase and decrease of Peptostreptococcus productus and Bacteroides vulgatus respectively, this non-significant decrease of Bacteroides vulgatus in asthma group is also in line with previous studies on diabetes type II patients and metabolic endotoxemia initiates obesity and insulin resistance, which showed non-significant decrease of Bacteroides vulgatus (Wu et al., 2010; Cani et al., 2007).
In human, the gut microbiota mainly divides into two groups, Firmicutes and Bacteroidetes (Xu et al., 2007). Bacteroides vulgatus is, numerically, predominant species in gut microbiota which indicates a complex but beneficial relationship with the host by prevention of intestinal colonization (Wells et al., 1988; Wexler, 2007). In our routine daily life, the most frequently used probiotics are mainly the Lactobacillus and Bifidobacterium genera and have health benefits (Butel, 2014). Bifidobacterium and Lactobacillus are constantly reduced in colorectal cancer (Borges-Canha et al., 2015) and they have also shown anti-atherogenic, anti-inflammatory and anti-obesity effects in number of studies (Nova et al., 2016). Different lactobacillus strains have similar moderate antimicrobial activities against pathogens (Shim et al., 2016). While, Peptostreptococcus productus, is a reductive acetogenic strain, with its continuous administration, competes with methanogens for H2 in the gut microbial ecosystem due to its reductive acetogenic character (Nollet et al., 1997).
Conclusion
In this study, we observed the change in characterization of gut microbiota of asthma patients. It also indicates that gut microbiota of asthma patients has some significant changes which is associated with disease occurrence. DGGE analysis shows that a significant change in gut microbial diversity and similarity in disease group as compared to healthy controls. Real time PCR results revealed that in asthma patients, Bacteroides vulgatus, Peptostreptococcus productus have exhibited the changes of different degree, while copy numbers of Bifidobacterium, Lactobacillus and Clostridium leptum sub group are significantly reduced in asthma group. So, more multicenter studies are required with larger sample size of both patients and healthy subjects to understand the process and mechanism of dysbiosis in gut microbiota with the development of asthma.
Acknowledgement
We would like to thank Dr. Muhammad Nawaz, Assistant Professor at UVAS, Pakistan for providing technical advice for completing the project.
Limitation of study
The smaller sample size is one of the limitations of our study and it is mainly because of the unwillingness of the patients to give their fecal samples. Moreover, excluding the patients and healthy subjects taking other drugs apart from antibiotics, could also improve our understanding about the subject matter.
Statement of conflict of interest
We hereby disclose that we do not have any conflict of interest including any financial, personal or other relationships with other people or organizations.
References
Bonamichi-Santos, R., Aun, M., Agondi, R., Kalil, J. and Giavina-Bianchi, P., 2015. Microbiome and asthma: What have experimental models already taught us? J. Immuunol. Res., 2015: Article ID 614758. http://dx.doi.org/10.1155/2015/614758
Borges-Canha, M., Portela-Cidade, J.P., Dinis-Ribeiro, M., Leite-Moreira, A.F. and Pimentel-Nunes, P., 2015. Role of colonic microbiota in colorectal carcinogenesis. Rev. Esp. Enfern. Dig., 107: 659-671. https://doi.org/10.17235/reed.2015.3830/2015
Butel, M.J., 2014. Probiotics, gut microbiota and health. Med. Mal. Infect., 44: 1-8. https://doi.org/10.1016/j.medmal.2013.10.002
Cani, P.D., Amar, J., Iglesias, M.A., Poggi, M., Knauf, C., Bastelica, D., Neyrinck, A.M., Fava, F., Tuohy, K.M., Chabo, C., Waget, A., Delmee, E., Cousin, B., Sulpice, T., Chamontin, B., Ferrieres, J., Tanti, J.F., Gibson, G.R., Casteilla, L., Delzenne, N.M., Alessi, M.C. and Burcelin, R., 2007. Metabolic endotoxemia initiates obesity and insulin resistance. Diabetes, 56: 1761-1772. https://doi.org/10.2337/db06-1491
Dorigo, U., Volatier, L. and Humbert, J.F., 2005. Molecular approaches to the assessment of biodiversity in aquatic microbial communities. Water Res., 39: 2207-2218. https://doi.org/10.1016/j.watres.2005.04.007
Dubernet, S., Desmasures, N. and Guéguen, M., 2002. A PCR-based method for identification of lactobacilli at the genus level. FEMS Microbiol. Lett., 214: 271-275. https://doi.org/10.1111/j.1574-6968.2002.tb11358.x
Fromin, N., Hamelin, J., Tarnawski, S., Roesti, D., Jourdain-Miserez, K., Forestier, N., Teyssier-Cuvelle, S., Gillet, F., Aragno, M. and Rossi, P., 2002. Statistical analysis of denaturing gel electrophoresis (DGE) fingerprinting patterns. Environ. Microbiol., 4: 634-643. https://doi.org/10.1046/j.1462-2920.2002.00358.x
Gafan, G.P., Lucas, V.S., Roberts, G.J., Petrie, A., Wilson, M. and Spratt, D.A., 2005. Statistical analyses of complex denaturing gradient gel electrophoresis profiles. J. clin. Microbiol., 43: 3971-3978. https://doi.org/10.1128/JCM.43.8.3971-3978.2005
Gill, S.R., Pop, M., Deboy, R.T., Eckburg, P.B., Turnbaugh, P.J., Samuel, B.S., Gordon, J.I., Relman, D.A., Fraser-Liggett, C.M. and Nelson, K.E., 2006. Metagenomic analysis of the human distal gut microbiome. Science., 312: 1355-1359. https://doi.org/10.1126/science.1124234
Green, S.J., Leigh, M.B. and Neufeld, J.D., 2010. Denaturing gradient gel electrophoresis (DGGE) for microbial community analysis. In: Handbook of hydrocarbon and lipid microbiology. Springer. https://doi.org/10.1007/978-3-540-77587-4_323
Guarner, F. and Malagelada, J.R., 2003. Gut flora in health and disease. The Lancet, 361: 512-519. https://doi.org/10.1016/S0140-6736(03)12489-0
Högenauer, C., Langner, C., Beubler, E., Lippe, I.T., Schicho, R., Gorkiewicz, G., Krause, R., Gerstgrasser, N., Krejs, G.J. and Hinterleitner, T.A., 2006. Klebsiella oxytoca as a causative organism of antibiotic-associated hemorrhagic colitis. N. Engl. J. Med., 355: 2418-2426. https://doi.org/10.1056/NEJMoa054765
Huang, Y.J. and Boushey, H.A., 2015. The microbiome in asthma. J. Allergy clin. Immunol., 135: 25-30. https://doi.org/10.1016/j.jaci.2014.11.011
Ishaq, H.M., Mohammad, I.S., Guo, H., Shahzad, M., Hou, Y.J., Ma, C., Naseem, Z., Wu, X., Shi, P. and Xu, J., 2017a. Molecular estimation of alteration in intestinal microbial composition in Hashimoto’s thyroiditis patients. Biomed. Pharmacother., 95: 865-874.
Ishaq, H.M., Mohammad, I.S., Guo, H., Shahzad, M., Hou, Y.J., Ma, C., Naseem, Z., Wu, X., Shi, P. and Xu, J., 2017b. Molecular characterization of fecal microbiota of healthy chinese tobacco smoker subjects in Shaanxi Province, Xi’an China. J. Ayub Med. Coll. Abbottabad, 29: 3–7.
Kabeerdoss, J., Sankaran, V., Pugazhendhi, S. and Ramakrishna, B.S., 2013. Clostridium leptum group bacteria abundance and diversity in the fecal microbiota of patients with inflammatory bowel disease: a case-control study in India. BMC Gastroenterol., 13: 20. https://doi.org/10.1186/1471-230X-13-20
Kamada, N., Chen, G.Y., Inohara, N. and Núñez, G., 2013. Control of pathogens and pathobionts by the gut microbiota. Nat. Immunol., 14: 685-690. https://doi.org/10.1038/ni.2608
Khoruts, A., Dicksved, J., Jansson, J.K. and Sadowsky, M.J., 2010. Changes in the composition of the human fecal microbiome after bacteriotherapy for recurrent Clostridium difficile-associated diarrhea. J. clin. Gastroenterol., 44: 354-360.
Ledder, R.G., Gilbert, P., Huws, S.A., Aarons, L., Ashley, M.P., Hull, P.S. and Mcbain, A.J., 2007. Molecular analysis of the subgingival microbiota in health and disease. Appl. environ. Microbiol., 73: 516-523. https://doi.org/10.1128/AEM.01419-06
Ley, R.E., Bäckhed, F., Turnbaugh, P., Lozupone, C.A., Knight, R.D. and Gordon, J.I., 2005. Obesity alters gut microbial ecology. Proc. natl. Acad. Sci. U.S.A., 102: 11070-11075. https://doi.org/10.1073/pnas.0504978102
Ling, Z., Kong, J., Liu, F., Zhu, H., Chen, X., Wang, Y., Li, L., Nelson, K.E., Xia, Y. and Xiang, C., 2010. Molecular analysis of the diversity of vaginal microbiota associated with bacterial vaginosis. BMC Genom., 11: 488. https://doi.org/10.1186/1471-2164-11-488
Lyons, S.R., Griffen, A.L. and Leys, E.J., 2000. Quantitative real-time PCR for Porphyromonas gingivalis and total bacteria. J. clin. Microbiol., 38: 2362-2365.
Ma, C., Wu, X., Nawaz, M., Li, J., Yu, P., Moore, J.E. and Xu, J., 2011. Molecular characterization of fecal microbiota in patients with viral diarrhea. Curr. Microbiol., 63: 259-266. https://doi.org/10.1007/s00284-011-9972-7
Matsuki, T., Watanabe, K., Fujimoto, J., Miyamoto, Y., Takada, T., Matsumoto, K., Oyaizu, H. and Tanaka, R., 2002. Development of 16S rRNA-gene-targeted group-specific primers for the detection and identification of predominant bacteria in human feces. Appl. environ. Microbiol., 68: 5445-5451. https://doi.org/10.1128/AEM.68.11.5445-5451.2002
Matsuki, T., Watanabe, K., Fujimoto, J., Takada, T. and Tanaka, R., 2004. Use of 16S rRNA gene-targeted group-specific primers for real-time PCR analysis of predominant bacteria in human feces. Appl. environ. Microbiol.,70: 7220-7228. https://doi.org/10.1128/AEM.70.12.7220-7228.2004
McBain, A.J., Bartolo, R.G., Catrenich, C.E., Charbonneau, D., Ledder, R.G., Rickard, A.H., Symmons, S.A. and Gilbert, P., 2003. Microbial characterization of biofilms in domestic drains and the establishment of stable biofilm microcosms. Appl. environ. Microbiol., 69: 177-185. https://doi.org/10.1128/AEM.69.1.177-185.2003
Muir, A.B., Benitez, A.J., Dods, K., Spergel, J.M. and Fillon, S.A., 2016. Microbiome and its impact on gastrointestinal atopy. Allergy, 71: 1256-1263. https://doi.org/10.1111/all.12943
Muyzer, G., de Waal, E.C. and Uitterlinden, A.G., 1993. Profiling of complex microbial populations by denaturing gradient gel electrophoresis analysis of polymerase chain reaction-amplified genes coding for 16S rRNA. Appl. environ. Microbiol., 59: 695-700.
Nollet, L., Velde, I.V. and Verstraete, W., 1997. Effect of the addition of Peptostreptococcus productus ATCC35244 on the gastro-intestinal microbiota and its activity, as simulated in an in vitro simulator of the human gastro-intestinal tract. Appl. environ. Microbiol., 48: 99-104. https://doi.org/10.1007/s002530051022
Nova, E., Perez de Heredia, F., Gomez-Martinez, S. and Marcos, A., 2016. The role of probiotics on the microbiota: Effect on obesity. Nutr. Clin. Pract., 3: 387-400. https://doi.org/10.1177/0884533615620350
Power, S.E., O’toole, P.W., Stanton, C., Ross, R.P. and Fitzgerald, G.F., 2013. Intestinal microbiota, diet and health. Br. J. Nutr., 111: 387-402. https://doi.org/10.1017/S0007114513002560
Scanlan, P.D., Shanahan, F., O’Mahony, C. and Marchesi, J.R., 2006. Culture-independent analyses of temporal variation of the dominant fecal microbiota and targeted bacterial subgroups in Crohn’s disease. J. clin. Microbiol., 44: 3980-3988. https://doi.org/10.1128/JCM.00312-06
Shim, Y.H., Lee, S.J. and Lee, J.W., 2016. Antimicrobial activities of Lactobacillus strains against uropathogens. Pediat. Int., 58: 1009-1013. https://doi.org/10.1111/ped.12949
Silva, E. and Russo, C., 2000. Techniques and statistical data analysis in molecular population genetics. Hydrobiologia, 420: 119-135. https://doi.org/10.1023/A:1003993824352
Tan, J., Mckenzie, C., Potamitis, M., Thorburn, A., Mackay, C. and Macia, L., 2013. The role of short-chain fatty acids in health and disease. Adv. Immunol., 121: 91-119. https://doi.org/10.1016/B978-0-12-800100-4.00003-9
van Der Gucht, K., Sabbe, K., de Meester, L., Vloemans, N., Zwart, G., Gillis, M. and Vyverman, W., 2001. Contrasting bacterioplankton community composition and seasonal dynamics in two neighbouring hypertrophic freshwater lakes. Environ. Microbiol., 3: 680-690. https://doi.org/10.1046/j.1462-2920.2001.00242.x
Walsh, C.J., Guinane, C.M., O’Toole, P.W. and Cotter, P.D., 2014. Beneficial modulation of the gut microbiota. FEBS Lett., 588: 4120-4130. https://doi.org/10.1016/j.febslet.2014.03.035
Wang, R.F., Cao, W.W. and Cerniglia, C.E., 1996. PCR detection and quantitation of predominant anaerobic bacteria in human and animal fecal samples. Appl. environ. Microbiol., 62: 1242-1247.
Wang, Z., Yang, W., Sun, L., Zhu, B., Li, D. and Liu, C., 2016. Characterization of a neutral protease gene of Bacillus subtilis isolated from the guts of Bombyx mori. Pakistan J. Zool., 48: 179-185.
Wells, C., Maddaus, M., Jechorek, R. and Simmons, R., 1988. Role of intestinal anaerobic bacteria in colonization resistance. Eur. J. clin. Microbiol. Infect. Dis., 7: 107-113. https://doi.org/10.1007/BF01962194
Wexler, H.M., 2007. Bacteroides: the good, the bad and the nitty-gritty. Clin. Microbiol. Rev., 20: 593-621. https://doi.org/10.1128/CMR.00008-07
Wu, X., Ma, C., Han, L., Nawaz, M., Gao, F., Zhang, X., Yu, P., Zhao, C.A., Li, L. and Zhou, A., 2010. Molecular characterisation of the faecal microbiota in patients with type II diabetes. Curr. Microbiol., 61: 69-78. https://doi.org/10.1007/s00284-010-9582-9
Xu, J., Mahowald, M.A., Ley, R.E., Lozupone, C.A., Hamady, M., Martens, E.C., Henrissat, B., Coutinho, P.M., Minx, P. and Latreille, P., 2007. Evolution of symbiotic bacteria in the distal human intestine. PLoS Biol., 5: e 0050156.
Zhou, L., Li, X., Ahmed, A., Wu, D., Liu, L., Qiu, J., Yan, Y., Jin, M. and Xin, Y., 2014. Gut microbe analysis between hyperthyroid and healthy individuals. Curr. Microbiol., 69: 675-680. https://doi.org/10.1007/s00284-014-0640-6
To share on other social networks, click on any share button. What are these?