
Advances in Animal and Veterinary Sciences


Research Article

Development of Recombinant Nucleocapsid Protein based indirect ELISA for Serodiagnosis of Peste des Petits Ruminants in Sheep and Goats
Vinayagamurthy Balamurugan1*, Manisha Roy1, Shashikumar Sowjanyakumari1, Sunil Abraham1, Rizvan Apsana1, Mohandoss Nagalingam1, Divakar Hemadri1, Habibur Rahman1,2
1Indian Council of Agricultural Research-National Institute of Veterinary Epidemiology and Disease Informatics (ICAR-NIVEDI), Ramagondanahalli, Post Box No. 6450, Yelahanka, Bengaluru- 560 064, Karnataka, India; 2Deputy Director General (Animal Science), Division of Animal Science, Krishi Bhavan, New Delhi - 110 001, India.
Abstract | In this study, expression of immunogenic portion of Peste des Petits ruminants virus (PPRV) nucleocapsid (N) protein in Escherichia coli (BL21) was envisaged to evaluate the potential use of recombinant protein as a diagnostic antigen in polyclonal antibodies based indirect ELISA for serodiagnosis. The immunogenic region of N gene coding sequences from PPR vaccine virus (Sungri 96 strain) was amplified, cloned in pET32a vector and expressed in E. coli as fusion protein for bulk production and easy Ni-NTA His-tag purification. The recombinant PPRV N protein (rPPRVNP) was expressed in E. coli at an optimal temperature of 37 °C with 1 mM IPTG for 5 h post induction and characterized by SDS-PAGE and western blot using PPRV-specific monoclonal and polyclonal antibodies or serum or anti-His-tag conjugate that confirmed PPRV specific protein with a size of ~50kDa. The expressed rPPRVNP was in insoluble form and was purified under denaturation condition by Ni-NTA purification method followed by refolding, renaturation methods and further concentrated by protein cut-off concentrators for obtaining single protein band. The rPPRVNP was assessed for its immunoreactivity as diagnostic antigen by immunoblotting and ELISA using standard PPRV specific antibodies. The immunogenic reactivity of expressed rPPRVNP was optimized in indirect ELISA using known true positive and negative sera with respect to PPRV antibodies. On standardization of rPPRVNP based indirect ELISA using 661serum samples, the relative diagnostic sensitivity and specificity of the assay 83.76% and 83.13% respectively was observed at cut off level of 25 Per cent Positivity (PP) with an agreement of Cohen’s kappa value of 0.648 with good agreement. This indirect ELISA as additional diagnostic tool for diagnosis of PPR in sheep and goats and rPPRVNP could be a sustainable source of safe antigen in countries of non-endemicity without the need to handle infectious virus for sero-diagnosis.
Keywords | PPR virus, Recombinant, nucleocapsid, E. coli, indirect ELISA, serodiagnosis
Editor | Kuldeep Dhama, Indian Veterinary Research Institute, Uttar Pradesh, India.
Received | May 27, 2016; Accepted | June 11, 2016; Published | June 16, 2016
*Correspondence | Dr. V. Balamurugan, M.V.Sc., Ph.D., Postdoc (Germany), FIVS, Senior Scientist, ICAR-NIVEDI, Ramagondanahalli, Post Box No. 6450, Yelahanka, Bengaluru- 560 064, Karnataka, India; Email: [email protected]; [email protected]
Citation | Balamurugan V, Roy M, Sowjanyakumari S, Abraham S, Apsana R, Nagalingam M, Hemadri D, Rahman H (2016). Development of recombinant Nucleocapsid protein based indirect ELISA for serodiagnosis of Peste des Petits ruminants in sheep and goats. Adv. Anim. Vet. Sci. 4(6): 301-310.
DOI | Http://dx.doi.org/10.14737/journal.aavs/2016/4.6.301.310
ISSN (Online) | 2307-8316; ISSN (Print) | 2309-3331
Copyright © 2016 Balamurugan et al. This is an open access article distributed under the Creative Commons Attribution License, which permits unrestricted use, distribution, and reproduction in any medium, provided the original work is properly cited.
INTRODUCTION
Peste des Petits ruminants (PPR) is a highly contagious viral disease of domestic and wild small ruminants and is currently emerging to cause infections in camels (Albina et al., 2013). The causative agent, PPR virus (PPRV) is a member of the genus Morbillivirus of the family Paramyxoviridae and contains approximately 16 kb long genome of a single-stranded negative sense RNA (Bailey et al., 2005). The disease was first described in 1942 in Côte d’Ivoire (Ivory Coast), West Africa, since then the disease has spread to different regions in sub-Saharan Africa, the Arabian Peninsula, the Middle East, Southwest Asia, India and other Asian countries (Parida et al., 2015). As of now, a total of 76 countries with approximately 1.7 billion sheep and goats population in Africa, the Middle East and the Indian subcontinent have confirmed PPR within their borders, and many countries are at risk of the disease being introduced (http://www.fao.org/ppr/en/).
In India, PPR was first reported from Arasur village, Villupuram, Tamil Nadu, during 1987 (Shaila et al., 1989). Since then, the disease became enzootic in many southern and northern states of India causing huge economic losses (Balamurugan et al., 2014). Now the disease is enzootic and outbreaks occurs throughout India almost all season of the year (Balamurugan et al., 2014; Muthuchelvan et al., 2015). Currently in India, sero-surveillance and/or sero-monitoring and clinical diagnosis of PPR is being carried out by monoclonal antibody (MAb) based competitive enzyme-linked immunosorbent assay (c-ELISA) (Singh et al., 2004b) and sandwich ELISA (s-ELISA) (Singh et al., 2004a) kits, respectively. However, the accidental loss of the monoclonal antibody-producing hybridoma clone owing to a laboratory accident or improper storage conditions (McCullough et al., 1986) constitutes a limitation of well-established MAb based assay some times. To accommodate such a situation in future, polyclonal antibody-based indirect ELISA has also been developed for the detection of PPRV antibodies for sero-epidemiological survey (Balamurugan et al., 2007).
In all these assays, cell culture attenuated live PPRV is being used as antigen. The bulk production of such virus antigen requires expertise and elaborate infrastructure. There is also possible batch to batch variation in antigen production and also involved in risk of handling live virus and accidental release into the environment. Therefore, generally, a recombinant antigen based diagnostic assay will be a better alternative not only for disease control and eradication but also useful for disease surveillance in the non-endemic countries and post-eradication phase where there could be restriction in handling of the live virus. Due to the advancement in rDNA and gene expression technologies, several attempts have been made since long to express various viral proteins in different expression systems and to assess potential use of recombinant proteins in various diagnostic assays.
Earlier recombinant antigen based diagnostic methods have been developed for the detection of several morbilliviruses such as rinderpest virus (RPV) (Kamata et al., 1993), PPRV (Ismail et al., 1995; Libeau et al., 1995; Choi et al., 2005b; Dechamma et al., 2006; Balamurugan et al., 2006; Yadav et al., 2009), canine distemper virus (CDV) (Latha et al., 2007) and Nipah virus (Yu et al., 2006). Among the structural proteins of morbilliviruses, N protein is the highly conserved immunogenic core protein (Lefebvre et al., 1991) and is expressed at a high level in infected cells (Diallo et al., 1994). Further, N protein has both type-specific and cross-reactive epitopes. So, it is a good antigen candidate for diagnosis of morbilliviruses. The nucleocapsid (N) protein is the most abundant, antigenically well conserved among morbilliviruses and is expressed to a very high level as compared to the other viral proteins, and is highly immunogenic while being located internally to the virus and can be used for diagnosis (Diallo, 1990; Bodjo et al., 2007).
Several researches have been used expressed PPRV N protein as diagnostic antigen in ELISA for serodiagnosis of the PPR. Earlier, recombinant baculovirus expressed PPRV N protein in insect cells was used as a coating antigen in ELISA for serodiagnosis of PPR. Further, Libeau et al. (1995) developed a c-ELISA based on the reaction between a MAb and a recombinant N protein of PPRV (Nigeria 75/1) expressed in baculovirus. Choi et al. (2005b) developed a rapid c-ELISA for diagnosis and surveillance of PPR. In addition, the expressed truncated and full-length N protein of PPRV in E. coli showed reactivity in s-ELISA and c-ELISA with MAbs (Yadav et al., 2009). Recently, Zhang et al. (2012) developed an indirect ELISA with artificially synthesized N protein of PPRV. Moreover, the amino-terminal domains of 420aa in the N protein of PPRV, are highly conserved across the genus and among PPRV strains. But, in RPV, it has been established that the epitopes at the amino-terminal half of the N protein are variable, highly immunogenic (Bodjo et al., 2007) and stimulate humoral immune response more rapidly than those in C-terminal region (Choi et al., 2005a). Prokaryotic E.coli system is used most commonly for protein which does not requires the post-translational modifications, since the expression is easy and it is possible to express the protein in bulk. Further, production of recombinant antigens in bacteria is simpler and more economical when compared to other heterologous system.
Keeping the recombinant protein could be a sustainable source of safe antigen in the diagnostic assays in countries of non-endemicity or in the post control and eradication phases of endemic countries without the need to handle infectious virus for serodiagnosis of disease, the present study, expression of immunogenic portion of PPRV N protein in E.coli was envisaged to evaluate the potential use of recombinant protein as a diagnostic antigen in a polyclonal antibody based indirect ELISA as additional diagnostic tool for diagnosis of PPR in sheep and goats.
MATERIALS AND METHODS
Amplification of PPRV N-Gene Sequences
The viral RNA was extracted from partially purified PPR vaccine virus (Sungri 96) by using RNeasy Mini Kit (Qiagen, Hilden, Germany) as per the manufacturer’s instructions. The extracted RNA was used as a template for reverse-transcription (RT) by using random hexamer primer (MBI, Fermentas, MD, USA) and 200U of MMuLV reverse transcriptase (Promega, Madison, USA). The amino terminal portion of N Protein coding sequence was amplified from the synthesized cDNA, by PCR using Pfu DNA polymerase (MBI, Fermentas, MD, USA) and PPRV N gene-specific primers PPRNF or (EcoRI): 5’-ATCTGAATTCATGGCTACTCTCCTTAAAAGC-3’ and PPRNRev (NotI) (His): 5’-ATG GCGGCCGCATGGTGATGGTGATGGTGGAGTCCGGCTTCGACAATATA-3’ modified from earlier reported one (Choi et al., 2005a; Yadav et al., 2009), which were designed based on published sequence (Accession #AY560591). The applied cycling conditions were: preheating at 95°C for 3min, 35 cycles of 94°C for 45sec, 50-60°C (incremental step up annealing +1°C/cycle) for 1 min and 72°C for 2 min with a final extension of 72°C for 10 min. The amplified products were analysed in agarose gel and documented.
Construction of Expression Cassette in pET32a Vector
The amplicon was purified from the agarose gel using GeneJET Gel Extraction Kit(Thermo Scientific Waltham, Massachusetts, USA) as per manufacturer’s instructions and directionally cloned in to pET32a vector at EcoRI and Not I sites after ligation and transformation into E.coli Top 10F’ competent cells (Invitrogen, Life technologies, CA, USA)by heat shock method. The recombinant transformed cells were plated on Luria Bertini (LB) agar containing ampicillin (50µg/ml), for selection of recombinant clones. The colonies (pET32a+ NP) were screened and confirmed by colony PCR, release of insert by restriction enzyme (RE) analysis and commercial DNA sequencing for identification of the cloned gene products in proper frame and orientation.
Expression of PPRV Immunogenic Portion of N Protein in E. coli
The confirmed recombinant plasmid DNA containing target gene in the proper frame in the vector backbone was transformed into E. coli BL21(DE3) pLysS (Invitrogen, Life technologies, CA, USA) and plated on LB agar containing ampicillin (50µg/ml) and Chloramphenicol (34µg/ml), for selection of recombinant BL-21 clones. The clones were screened for the presence of PPRV Ngene specific sequences by colony PCR. The positive individual colonies were grown at 37°C till the culture reached mid-log phase (OD 600nm of 0.4–0.5) and induced for expression of the cloned gene products. The expressed protein was characterized by Sodium Dodecyl Sulfate-Polyacrylamide Gel Electrophoresis (SDS-PAGE) and immunoblot /Western blot as per standard procedures described earlier.
To optimize the induction time for expression of cloned gene products (recombinant protein), the cultures were harvested at hourly intervals post induction (0, 1, 2, 3, 4, 5, 6, 7, 8, and 9 hours) under standard conditions of 1mM IPTG concentration and 37°C induction temperature. Similarly the optimal working concentration of IPTG (0.5, 1, 1.5 and 2 mM IPTG) and incubation temperature (18, 25, 30 and 37°C) were determined. The harvested induced cultures pellets were then mixed with sample loading buffer (v/v: 1/1, Sigma) heated at 100°C for 10 min for SDS–PAGE analysis.
Purification of Expressed Recombinant Protein
Harvested 50ml of induced culture pellet at 5 h post induction was suspended in 8ml urea lysis buffer, vortexed and incubated for an hour and supernatant was collected for Ni-NTA purification methods. The supernatant was allowed to bind with Ni-NTA Agarose (Genetix Nucleopore) for 4 h with gentle agitation. The suspension was passed through agarose column (Thermo Scientific Waltham, Massachusetts, USA) and flow through was collected. Further the column was washed with solublisation buffer (20mM TrisHcl, 1mM EDTA,500mM NaCl, 8M Urea, 0.5% Triton X-100, and 5mM β Mercaptoethanol) and protein was eluted with solublisation buffer containing different concentration of 100, 200, 300, 400 and 500mM imidazole.
The eluted protein was dialysed against dialysis buffer containing different concentration of urea in the decreasing concentration (8, 6, 4, 3, 2, 1 and 0 M with 2 h interval for each dialysis) for refolding or renaturation of purified denatured protein in order to bring the protein in solubilizing form. The dialyzed protein was further subjected to 30kDa and 50kDa protein cut-off concentrators (AmiconUltra filter, Merk Millipore) to obtain the single purified and concentrated PPRVNP for used as diagnostic antigen. The final concentration of purified single band PPRV NP was estimated by Bradford method. The rPPRVNP with protease inhibitors was stored in -80°C in aliquot and used as diagnostic antigen in ELISA as and when required.
SDS-Page and Western Blotting
The harvested post-induced recombinant bacteria samples, mock vector transformed bacteria, host bacteria and eluted fractions were mixed separately with SDS sample buffer and resolved by polyacrylamide gel (5% stacking and 12% resolving). The resolved proteins by SDS-PAGE were transferred on to a nitrocellulose membrane (NCM) (Sigma Aldrich, St. Louis, Missouri United States) following method described earlier (Yadav et al., 2009) and the membrane was blocked with 5% skimmed milk powder and 3% Lactalbumin Hydrolysate in PBS-Tween 20 buffer(pH7.2) overnight at 4°C. The blot was incubated with primary antibodies either PPRV specific-polyclonal serum or monoclonal antibodies (MAb) (s-ELIAS Kit - Singh et al., 2004a) or negative control serum or PPRV specific hyper immune serum and followed by incubation with secondary antibodies either anti-His-tag conjugate or anti-goat HRPO conjugate or anti-mouse HRPO conjugate (Sigma Aldrich, St. Louis, Missouri United States)as the case may be, and incubated for 1 h followed by washing of membrane and development of blot with Diaminobenzidine (DAB) as a chromogen and documentation.
Indirect ELISA Standardization
ELISA was carried out according to Balamurugan et al. (2007) with some modifications. Recombinant purified rPPRVNP was coated (in 50μl volume/well)in a flat bottomed 96 well plate (Nalgene Nunc Int., Hamburg, Germany) and stored in 4°C for overnight. The plate was further incubated at 37°C for 1h (optional), then washed thrice with PBST( 0.1% Tween-20) and blocked with 100μl of blocking buffer [1X PBST with 5% w/v Skim Milk Powder, 3% w/v lactalbumin Hydrolysate (pH 7.2)] for 1h at 37°C in a shaker incubator. All control sera (Strong positive, positive and negative) and test sera were diluted in 1:10 dilution in blocking buffer and added in 50μl volume/well. After incubation at 37°C for 1h, the plate were washed thrice with PBST (0.1% Tween-20). The antigen–antibody reaction was followed by incubation with anti-Sheep and Goat HRPO conjugate (1:3000 dilution) and detected by colour development with the chromogen, ortho-phenyl diamine (OPD) and H2O2 as the substrate. Substrate solution (OPD 1 mg/ ml containing 4 μl 3% H2O2) was added in each well and the colour reaction was developed for 15 min before stopping the reaction with 1 mol/L H2SO4 The absorbance values were measured at a wavelength of 492 nm. The assay included standard controls (conjugate, strong positive, and negative) to assess the quality and accuracy.
A checkerboard titration was performed for optimization of working dilutions of antigen and antibodies as per standard protocols. Antigen concentrations of 50ng to 500ng (50, 100, 200, 300, 400 and 500ng) and antibody dilutions of 6.25 to 200 (6.25, 12.5, 25, 50, 100 and 200) were performed for optimization of assay. The antigen and serum dilutions that gave maximum difference in absorbance at 492nm between positive and negative (P/N) were selected as strong positive and negative for further standardization.
For standardization of rPPRVNP based indirect ELISA, the serum sample from animals of varied immunological status (pre-vaccinated, post-vaccinated, infected or recovered animals) collected during field visit and outbreaks investigation were included in the study for initial screening of theses samples by PPR c-ELISA kit (Singh et al., 2004b) and PPR indirect ELISA (Balamurugan et al., 2007) for the selection of the true positive and true negative serum samples with respect to PPRV antibodies in order to determine cut-off value for the recombinant antigen based indirect ELISA for sheep and goats.
Statistical Analysis
The optimum combination of diagnostic specificity/sensitivity (Thrushfield, 2005) and cut off percentage positivity (PP) value were determined by frequency analysis graph (Wright et al., 1993). The raw data (OD values) obtained were converted into PP, using the following formula:
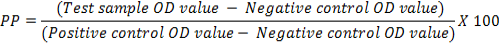
The cut off PP was determined from the frequency distribution of graph plotted with PP values obtained from 661 field samples. Cohen’s Kappa Test (Cohen, 1960) was used to determine a agreement. The Cohen’s Kappa coefficient was determined using open source software Win Episcope Version 2.0. Cut of value was derived from percent positivity (PP) by two sided contingency table from Receiver Operating Characteristics (ROC) curve. Sensitivity and Specificity of standardized ELISA was determined using the statistical formula given by Thrusfield (2005) using Vassar Stats online software.
RESULTS
Expression Cassette in pET32a Vector
The immunogenic region of N gene coding sequences from PPR vaccine virus was amplified, cloned in pET32a vector and expressed in E.coli as fusion protein. The partial 5’ end of PPRVN gene sequences was amplified by RT-PCR, which resulted in specific products of 838bp. The amplicons were ligated into thepET32a vector at EcoRI and NotI sites and ligated mixture were transformed in the E. coli TOP 10F’cells. The efficiency of transformation was 3×104/µg of ligated DNA products. The presence of the insert in the recombinant colonies was confirmed by colony PCR and RE analysis. Amplification of the gene specific product with virus specific primers, confirmed the presence of the insert five out of seven colonies screened. The positive colonies were grown and plasmid DNA was purified and subjected to RE digestion, which resulted in the release of cloned gene products from the recombinant clone and further nucleotide sequence of the insert in the recombinant clone was confirmed by sequencing which showed 100% identity of sequences of PPRV N gene of Sungri 96 strains.
Expression and Purification of rPPRVNP Protein in E. coli
In order to express PPRV NP, the recombinant cloned DNA was transformed into BL21 strain of E. coli by the heat-shock method along with the pET32a vector control.
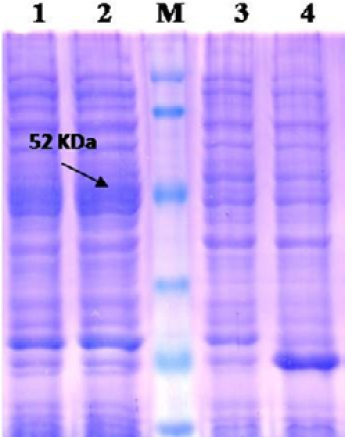
Lane 1: Insoluble fraction of cell lysate of recombinant NP clone 2; Lane 2: Soluble fraction of cell lysate of recombinant NP clone 2; Lane M: Prestained protein marker (Thermo Scientific 26612); Lane 3: BL-21 Host cell control; Lane 4: BL-21 –PET 32a Vector control.
The recombinant BL21 (DE3) pLysS clones were screened for presence of the target gene by PCR using insert specific primers, which result in amplification in six out of seven colonies screened. PCR positive BL21 clones were selected and induced to express PPRV NP with IPTG. On analysis of the expressed protein in SDS-PAGE, a single intense band of ~50 kDa could be seen corresponding to the expected expressed immunogenic PPRVNP (Figure 1), whereas no such protein bands were seen in the controls.
For better expression of over expressed PPRVNP clones, the expression conditions was optimized for various parameters, which showed the optimum induction time of 5h with 1mM IPTG induction at 37°C temperature. The expressed recombinant PPRV N Protein (rPPRVNP) was characterized by SDS-PAGE and western blot using PPRV- specific polyclonal and monoclonal serum/antibodies, anti-His-tag conjugate that confirmed the band with a ~ 50kDa size was PPRV specific protein, which was expressed in insoluble form. The rPPRVNP immunoblot with aforesaid primary antibodies showed an intense color protein corresponding to a ~50kDa expressed band (Figure 2).
Further protein was purified under denaturation condition by Ni-NTA purification method followed by refolding with different concentration of urea and obtained the single purified expressed protein by protein cut-off concentrators. The solubilisation buffer for elution of protein inNi-NTA agarose was optimized with 300mM imidazole for maximum yield (Figure 3). There were no contaminating
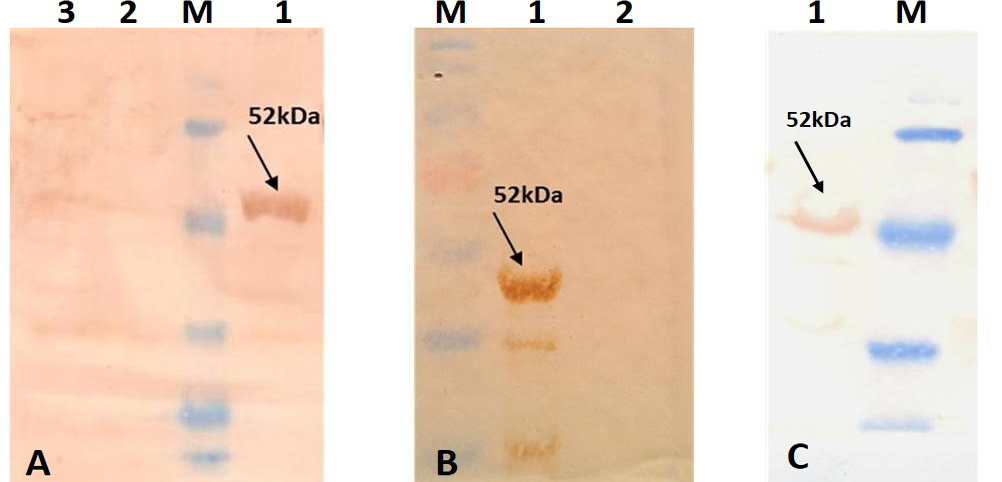
A) Immunoblotting with standard strong positive serum provided in the PPR c-ELISA kit; B) Immunoblotting with PPRV hyper immune serum; C) Immunoblotting with Anti-His tag conjugate (Pierce). Lane 1: Purified recombinant PPRV N Protein; Lane 2: BL-21 –PET 32a Vector control; Lane 3: BL-21 Host cell control; Lane M: Prestained protein Marker (Thermo Scientific 26612)
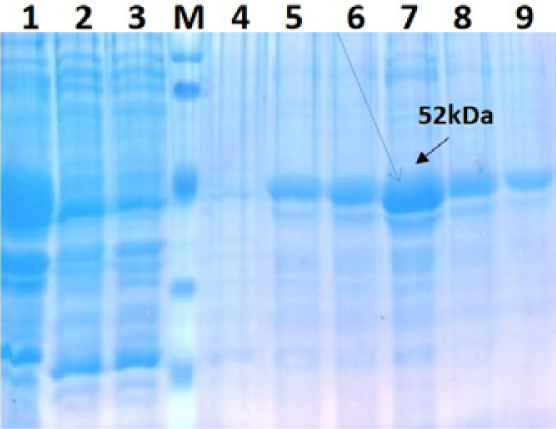
Lane 1: Insoluble fraction of cell lysate of recombinant NP clone 2; Lane 2: Flow Through; Lane 3 –Wash through; Lane M: Prestained protein marker (Thermo Scientific 26612); Lane 4: 9- Protein Elution with 0, 100, 200,300,400 and 500 mM Imidazole concentration
proteins in the separation and the concentration of purified rPPRVNP was 6mg/100ml of induced cell cultural pellet.
rPPRVNP Protein based Indirect ELISA
The immunogenicity of expressed protein was assessed by using PPRV- specific antibodies from sheep and goats, which showed reactivity with rPPRVNP in ELISA. Optimized antigen concentration of 200ng/well/50µl with serum dilution of 1:10 was selected along with conjugate dilution of 1:3000 was standardized for the assay (Figure 4). The optimum reactivity of the rPPRVNP was assessed using known positive (n=268) and negative (n=393) serum samples with respect to PPRV antibodies in c-ELISA. Sensitivity and specificity were calulated for 20 different cut-off values (5 to 100) by two sided contingency table using Receiver operative curve (ROC) curve cut off ≥ 25PP was arrived. The relative sensitivity and specificity of the assay with c-ELISA was 83.76% (78.26 to 88.11% at 95% CI) and 83.13% (79.16 to 86.49% at 95% CI) respectively with the Positive Predictive Value of 73.13 % (67.33 to 78.25 % at 95% CI) and Negative Predictive value of 90.33% (86.86 to 92.98 % at 95% CI) while the Cohen’s kappa value of 0.648 with good agreement.
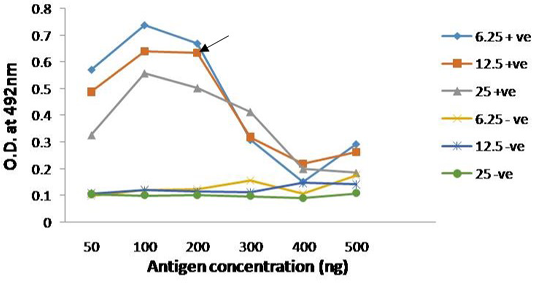
Figure 4: Reactivity of serially diluted positive serum with recombinant PPRV N Protein; Head of arrow indicates 75% absorbance (A492) of the plateau, which corresponds to optimal working dilution of the PPRV N protein and PPRV antibodies
DISCUSSION
PPR is considered as one of the main constraints in augmenting the productivity of small ruminants in endemic countries and chance of spread to other non-endemic countries due to trade or illegal migration of small ruminants populations in the border of the countries. The disease is economically important and hence, its control and eradication is a most priority. Considering the importance of small ruminants in food security and socio-economic growth mainly in Africa, Asia and in many other parts of the world, the complete control and eradication of PPR becomes essential and in this direction, recently, Food and Agricultural Organisation (FAO) and the World Organisation for Animal Health (OIE) convened in Abidjan, Côte d’Ivoire for the International Conference for the Control and Eradication of PPR, the high-level authorities from affected countries agreed on a global plan to control and eradicate PPR by 2030 (http://www.fao.org/news/story/ en/item/282397/icode/; Parida et al., 2015). This campaign will make PPR only the second animal disease ever to be eradicated, after rinderpest.
Use of whole virus antigen in diagnostic assay is not safe always, as it requires bio-safety measures and proper disposal, especially in countries of non-endemicity or in the post control and eradication phases, which warrants without the need to handle infectious virus for serological diagnostic assays. Hence, it is necessary to have a safe, potent and cost-effective antigen based diagnostics. Therefore, in the present study, expression of immunogenic portion (262aa from N-terminal region) of PPRVNP was expressed as ~50kDa recombinant fusion protein in E.coli system was envisaged to evaluate the potential use of recombinant protein as a diagnostic antigen with the development of polyclonal antibodies based indirect ELISA for serodiagnosis of PPR in sheep and goats.
The 5’ end of PPRV N gene sequences was amplified by RT-PCR, cloned into pET32a vector and characterized. In order to express PPRV NP, BL21 (DE3) pLysS strain of E. coli was used for transformation of the characterized recombinant plasmid DNA. After the characterization of the BL21 clones, expression of rPPRVNP was induced using 1mM IPTG at 37°C. Pre-induction incubation for 3h at 37°C was necessary to achieve mid-log phase growth. On comparison of protein profiles of the recombinant clones with that of the control (vector and BL21), molecular weight of ~50kDa expressed protein band was observed in case of recombinant pET32a+NP clone, as early as 4 h post induction (pi). The intensity of the bands increased gradually up to 7hpi. In un-induced and control cultures, such specific bands were not observed even after prolonged incubation. The optimum time of harvest was 5hpi and not much difference was observed in the rate of expression either at 6 or 7hpi but thereafter, there was a reduction in expression. This reduction could be attributed to autolysis of bacterial cells as reported earlier (Yadav et al., 2009). The proteins resolved in SDS-PAGE and transferred on to a nitro cellulose membrane and were detected by using a PPRV specific serum or antibodies. On analysis, ~50kDa molecular size was observed and confirmed that the expressed recombinant protein was specific to PPRV and no other non-specific bands were noticed. The predicted size of the expressed protein from amino acid sequences (32kDa) (Muthuchelvan et al., 2006) along with fusion His-tag (18kDa) is around ~50kDa. The calculated size as per aa composition along with fused His-tag and as observed by the mobility in SDS-PAGE, was in agreement with the reported size indicating that the 50kDa protein was the product from the cloned gene sequences. Western blot analysis with MAb against the PPRVNP/polyclonal antibodies or serum indicated that the bands observed in SDS-PAGE are virus specific. Such His-tag fusion protein has been expressed in case of PPR and Nipah virus also (Yu et al., 2006; Yadav et al., 2009).
Further, to assess the utility of the expressed protein, they were purified by using Ni-NTA affinity columns to their homogeneity. This method has been successfully used for purification of several other expressed proteins (Yu et al., 2006; Sun et al., 2007; Yadav et al., 2009). Presence of N-terminal His-tag in the vector as well as C-terminal His-tag both in cloned gene product and expression vector facilitated easy and efficient Ni-NTA column protein purification. It was found that 300mM imidazole concentration was optimum for elution of protein. However, various concentrations of imidazole (100–500mM) have been reported in purifying expressed protein (Yu et al., 2006; Latha et al., 2007; Pathak et al., 2008; Yadav, et al., 2009). Generally, purification under non-denaturing conditions avoids re-folding of denatured protein. Moreover, in this study, purification under denaturing conditions followed by re-folding of denatured protein in urea, resulted in good yield of protein in native solubilisation form and it was reacted in the immune assays and blot. This is advantageous to retain conformational epitopes on the protein and thereby facilitates better reactivity even with MAbs. It also helps to retain the immunogenicity of recombinant protein, as was reported earlier (Finzi et al., 2003).
Though other immune assays are used, ELISA is one of the most sensitive and extensively applied methods to evaluate expressed recombinant protein (Choi et al., 2005b; Li et al., 2006).The expressed protein was tested for its suitability as diagnostic antigen in indirect ELISA being used as a coating antigen for development of polyclonal antibodies based assay. Recombinant protein reactivity in ELISA indicated that the expressed PPRVNP reacted well with standard PPR serum in ELISA making it evident that the epitopes present in expressed protein are well recognized by the antibody. The OD values of all the standards used in this assay did not show much variation between the plates, indicating repeatability of the assay.
In order to determine the relative specificity and sensitivity of the assay while standardization, known positive and known negative samples with respect to PPRV antibodies were selected based on the preliminary screening of the serum samples by PPRV antigen based c-ELISA (Singh et al., 2004b), of which 268 positive and 393 negative samples were selected for determining the efficacy of the developed assay. A cut-off value (PP) of 25% was fixed upon comparison with established ELISA by two sided contingency table and ROC curve. The relative sensitivity and specificity of the assay was 83.76% and 83.13% respectively while the Cohen’s kappa value of 0.648 with good agreement.
However, when preliminary testing of the samples (n=218) showed the sensitivity (Dsn) and specificity (Dsp) of 84.29 and 89.69 % respectively. Initially, we thought, when testing with more number of serum samples, the sensitivity and specificity will increase. But, in a real situation, it was decreased when the samples size was increased to 661. This variation in the DSn and DSp may be due to the kinetics of the antibodies against H and N proteins of PPRV present in the serum samples of animals in various stages of infected or vaccinated animals in the field conditions. Moreover, in our earlier experimental study (personal communication) using periodically collected serum samples from few infected and vaccinated animals when tested with N and H protein based PPRV MAb based C-ELISA (Singh et al., 2004b; Yadav et al., 2009), it was observed that in the infected animals PPRV N specific antibodies appears earlier up to 10-14 days and afterwards PPRV H protein antibodies appears in the serum. Where as in vaccinated animals H protein specific antibodies appears earlier up to 14 days thereafter N protein specific antibodies. This needs further studies to be undertaken.
PPR free countries or countries which are at risk of PPR introduction may not accept the currently being used live attenuated PPRV antigen based ELISA kit. In these circumstances, the rPPRVNP polyclonal antibody based indirect ELISA would be the most sought assay as compared to whole PPRV antigen based kits. Further, it is well known that the prokaryotic expression system is cost-effective, user-friendly and easy to scale up commercially. It is also possible to produce the recombinant antigen on a large-scale in a single batch with use of fermenter. Such a product would have the homogeneity due to a single-cycle of down-stream processing. This indirect ELISA as additional diagnostic tool for diagnosis of PPR in sheep and goats and rPPRVNP could be a sustainable source of safe antigen in countries of non-endemicity without the need to handle infectious virus for sero-diagnosis.
ACKNOWLEDGEMENTS
Authors wish to thank Indian Council of Agricultural Research (ICAR), New Delhi, India, for constant support and encouragement. The financial support from Department of Biotechnology, GOI, in the form of project work (BT/PR/3482/ADV/90/121/2011) is gratefully acknowledged. The authors also thank staff of NIVEDI for providing their constant support and kind co-operation. Authors also wish to thank Department of Animal Husbandry and Veterinary Sciences and field Veterinarians for the kind help rendered during outbreak investigations and samples collections.
CONFLICT OF INTEREST
No conflicts of interests are declared by authors for the contents in the manuscript.
AUTHORS’ CONTRIBUTION
V. Balamurugan designed and carried out the experiment, interpreted the data and wrote the draft of manuscript. M. Roy, S. Sowjanyakumari, S. Abraham, R. Apsana, carried out the experiment. M. Nagalingam, performed the statistics, interpreted the data and provided support and edited manuscript. D. Hemadri, and H. Rahman provided guidance and support to carry out the experiments.
REFERENCES

