
Advances in Animal and Veterinary Sciences


Research Article

Detection and Typing of CPV with Real-Time PCR and Mini-Sequencing
Hariprasad Naidu Gonuguntla1,2, Kota Sri Naga Leela Surendra1, Samir Kumar Rana1, Nadikerianda Muthappa Ponnanna1, Bhaskaran Mohana Subramanian3, Girish Kumar Sharma4, Villuppanoor Alwar Srinivasan5*
1National Dairy Development Board, R&D Laboratory, Hyderabad, 500032, Telangana, India; 2Department of Biotechnology, Acharya Nagarjuna University, Guntur, Andhra Pradesh, 522004, India; 3Translational Research Platform for Veterinary Biologicals, Tamil Nadu Veterinary and Animal Sciences University, Chennai, 600051, India; 4National Dairy Development Board, Anand, 388001, Gujarat, India; 5National Dairy Development Board, Advisor, Animal Health, 33, Telecom Nagar, Gachibowli, Hyderabad 500032, Telangana, India.
Abstract | Canine parvovirus causes an acute enteric disease in dogs. All the infections currently reported, world-wide, are caused by either of the genetic variants of CPV2 viz., CPV2a, CPv2b and CPV2c. This study describes a novel, combinatorial approach for rapid and direct detection of CPV2 from canine fecal samples followed by typing to determine the variants (CPV2a, CPV2b and CPV2c) involved in the infection. The combined approach adapts a SYBR green based real-time PCR assay for the detection followed by the mini-sequencing assay of the resultant PCR amplicons for the variant typing. The real-time PCR assay reported here was standardized and validated with plasmid constructs bearing target DNA of CPV2a, 2b, 2c; and CPV-2b genome as source of templates. The assay could detect all three variants of CPV2 with a lower limit of detection (LOD) of one copy per reaction. Subsequently, the real-time PCR with viral genomic DNA extracted from tissue culture fluid (TCF) post viral culture showed a LOD of 5fg of genomic DNA per reaction. Melt-cure analysis of the reactions showed specific amplification of a ~145bp length fragment (Tm = ~73.3°C) matching the size of the target DNA. Further, reaction set-up with genomic DNA from unrelated viruses which causes infection in canines showed no amplified product. Together, it indicates the specificity of the assay. Downstream analysis of the PCR amplicons with the mini-sequencing method could adequately determine the variants at an initial concentration of one copy of plasmid construct or 5.0fg of genomic DNA. In a field study, of the fecal samples (n=60) collected from dogs showing enteric symptoms, 5, 2, 14 and 22 numbers of samples were determined positive by the virus isolation, hemagglutination (HA), PCR and real-time PCR assays, respectively. Samples determined positive by real-time PCR (n=22) were further genotyped by mini-sequencing. and all were found to be of CPV type 2a. The combined approach of real-time PCR followed by mini-sequencing for detection and typing respectively is specific and rapid. We reckon, this approach has the potential for extensive on-field applications in routine diagnosis and epidemiology.
Keywords | CPV, Real-time PCR, Mini-sequencing, CPV typing
Editor | Kuldeep Dhama, Indian Veterinary Research Institute, Uttar Pradesh, India.
Received | April 04, 2016; Accepted | April 09, 2016; Published | April 27, 2016
*Correspondence | Dr. Villuppanoor Alwar Srinivasan, Advisor (Animal Health), National Dairy Development Board, 33, Telecom Nagar, Gachibowli, Hyderabad 500032, India; E-mail: [email protected]
Citation | Gonuguntla HN, Surendra KSNL, Rana SK, Ponnanna NM, Subramanian BM, Sharma GK, Srinivasan VA (2016). Detection and typing of CPV with real-time PCR and mini sequencing. Adv. Anim. Vet. Sci. 4(4): 187-194.
DOI | Http://dx.doi.org/10.14737/journal.aavs/2016/4.4.187.194
ISSN (Online) | 2307-8316; ISSN (Print) | 2309-3331
Copyright © 2016 Gonuguntla et al. This is an open access article distributed under the Creative Commons Attribution License, which permits unrestricted use, distribution, and reproduction in any medium, provided the original work is properly cited.
INTRODUCTION
Canine parvovirus (CPV) is classified under the family Parvoviridae. Infection of dogs with CPV type 2 (CPV2) is characterized by a loss of appetite, vomiting and severe diarrhea. The phylogenetic and evolutionary studies suggest that CPV2 has originated from the genetically close and antigenically related feline panleucopenia virus (FPLV; Mochizuki M et al., 1991).
CPV2 genome comprises of a single stranded DNA molecule that encodes two structural proteins VP1 and VP2 and two non-structural proteins NS1 and NS2. VP2 protein binds to the transferrin receptor in the host cells which facilitates the entry of the virus (Hafenstein et al., 2007; Palermo et al., 2003, 2006; Hueffer et al., 2003).Nucleotide substitutions in the VP2 gene has led to the emergence of new variants of CPV2 (Martella et al., 2006). To date three variants namely CPV2a, CPV2b and CPV2c that differ genetically and antigenically from CPV2 have been documented from various countries (Parrish et al., 1985, 1991; Buonavoglia et al., 2001; Nakamura et al., 2004; Decaro et al., 2005a; Chinchkar et al., 2006). Surveillance tools for accurate identification of the CPV2 variants involved in infection are necessary for the adoption of suitable prophylaxis and control of the disease.
Conventional methods of diagnosis viz., haemagglutination inhibition assay (HI), virus isolation (VI) by cell culture, indirect fluorescent antibody test (IFAT) and polymerase chain reaction (PCR) fail to identify the variants causing the infection or disease (Mochizuki et al., 1993b; Senda et al., 1995; Pereira et al., 2000). However, real-time PCR using Minor groove binding (MGB) probes targeting the single nucleotide polymorphisms (SNPs) in VP2 gene can differentiate the CPV variants involved in infection. Besides, the real-time PCR method has been shown to be more sensitive than conventional PCR for the detection of viral DNA in faecal samples of dogs (Decaro et al., 2005c). Hence it has been used to study the prevalence of the virus and efficacy of the vaccine (Decaro et al., 2005b).
This laboratory had previously described a mini-sequencing based approach for differentiating CPV variants (Naidu et al., 2012). However, the method is does not quantify the load of CPV virus in clinical samples. The present study describes the development and evaluation of a rapid method that combines real-time PCR and mini-sequencing for the detection, quantification as well as typing of CPV2 variants namely 2a, 2b and 2c.
MATERIALS AND METHODS
Samples
Rectal swabs (n=60) were collected from the dogs showing clinical symptoms of fever and diarrhea in and around Hyderabad. Post collection the swabs were transported in 2 ml of transport medium (sterile phosphate buffer saline (PBS)) in 5 ml vials to the laboratory in cool packs.
Sample Processing
The swabs were vortexed briefly and the sample was transferred to 1.5 ml tube and centrifuged at 4,000 rpm for 5 minutes. The supernatant was then filtered through 0.2µ syringe filter and the filtrate thus obtained was examined by PCR (Senda et al., 1995), real-time PCR and HA test as described by Carmichael et al. (1980) for the presence of CPV. For the samples showing the presence of CPV, mini-sequencing reaction was performed.
Plasmid Constructs
Plasmid constructs viz., TOPO-CPV2a, TOPO-CPV2b and TOPO-CPV2c; (Naidu et al., 2012) harboring a 576 bp region (in between 3622bp and 4197bp of CPV genome; GenBank accession number M38245) of VP2 gene of CPV2 variants 2a, 2b and 2c respectively were used as reference controls in the present study.
Real-time PCR
The following primers viz., Forward primer - qCPV-145-F 5’-CACAACAGGAGAAACACCTGAG-3’, Reverse primer - qCPV-145-R 5’-CCAATTGGATCTGTTGGTAGC-3’ specific to the highly conserved regions of CPV VP2 gene were used in the real time PCR. The primers were designed using Primer3 plus online tool. The real-time PCR with the above primers amplified a 145 bp product in between 3952 bp and 4095 bp of CPV genome (GenBank accession number AJ564427) that corresponds to a region within the 576 bp portion (as in the Plasmid Constructs ) of the VP2 gene. All reactions were performed in 25 µl reaction volumes with the following constituents 12.5 µl of SYBR green real-time PCR master mix (Applied Biosystems), 5 picomoles of forward and reverse primers, and 5 µl of template DNA. The thermal cycling conditions were as follows- Initial denaturation at 95°C for 10 minutes; 40 cycles of denaturation at 94°C for 15sec; annealing and extension at 60°C for 1 min. Melting curve analysis was performed as a routine QC procedure for the real-time PCR assays.
Analytical Sensitivity of Real-time PCR
The sensitivity, as the minimum number of plasmid copies that could be detected in the real-time PCR assay was determined using the TOPO-CPV2b plasmid construct. Ten-fold serial dilutions of TOPO-CPV2b with a starting concentration of 0.2x1010 copies/5µl to 0.2 x10-3 copies/µl were prepared. Real-time PCR assay was performed for each dilution of TOPO-CPV2b (5 µl of each dilution per reaction i.e 1010 to 10-3 copies) in triplicates. Mean threshold cycle (Ct/ Cq) values, standard deviation (SD), standard error (SE), confidence interval (CI) and co-efficient of variation (CV) for each dilution of the constructs were estimated. The lower limit of detection (LOD) and lower limit of quantification (LOQ) in plasmid copy numbers were determined.
Similarly the sensitivity of the assay in minimum concentration (grams) of genomic DNA was determined using
Table 1: Statistical attributes of the plasmid and genomic DNA amplification performed in triplicates using SYBR green real-time PCR
|
S No |
Sample ID |
Quantity |
Cq (1) |
Cq (2) |
Cq (3) |
SD |
Mean |
SE |
CI (at 95%) |
|
1 |
S1 |
1.00E+07 |
15.6 |
15.4 |
15.4 |
0.1 |
15.4 |
0.051 |
0.11 |
|
2 |
S2 |
1.00E+06 |
18.2 |
18 |
19.5 |
0.79 |
18.5 |
0.395 |
0.89 |
|
3 |
S3 |
1.00E+05 |
21.6 |
21.3 |
21.1 |
0.29 |
21.3 |
0.146 |
0.33 |
|
4 |
S4 |
1.00E+04 |
25 |
24.7 |
24.2 |
0.39 |
24.6 |
0.194 |
0.44 |
|
5 |
S5 |
1.00E+03 |
28.8 |
28.5 |
28.5 |
0.16 |
28.6 |
0.081 |
0.18 |
|
6 |
S6 |
1.00E+02 |
32.2 |
32.3 |
31.8 |
0.27 |
32.1 |
0.134 |
0.31 |
|
7 |
S7 |
1.00E+01 |
36 |
34.5 |
36.1 |
0.94 |
35.5 |
0.469 |
1.06 |
|
8 |
G1 |
5.00E-02 |
9.7 |
9.6 |
9.7 |
0.04 |
9.7 |
0.019 |
0.05 |
|
9 |
G2 |
5.00E-03 |
13.4 |
13.1 |
13.1 |
0.14 |
13.2 |
0.069 |
0.16 |
|
10 |
G3 |
5.00E-04 |
16.9 |
16.5 |
16.4 |
0.27 |
16.6 |
0.133 |
0.31 |
|
11 |
G4 |
5.00E-05 |
20.4 |
20.4 |
19.9 |
0.33 |
20.2 |
0.164 |
0.37 |
|
12 |
G5 |
5.00E-06 |
24.1 |
24 |
23.5 |
0.34 |
23.9 |
0.171 |
0.38 |
|
13 |
G6 |
5.00E-07 |
27.7 |
27.3 |
27.2 |
0.3 |
27.4 |
0.151 |
0.34 |
|
14 |
G7 |
5.00E-08 |
31.5 |
31.5 |
31 |
0.27 |
31.3 |
0.133 |
0.31 |
Serial Number 1 to 7 are tenfold dilutions of positive plasmid standard (S1 represents 1.00E+07copies/reaction) Serial No 8 to 14 are genomic DNA standards varied by tenfold from preceding sample (G1 represents the Genomic DNA of 5ng/ reaction)
the genomic DNA extracted from CPV type 2b cultured in A72 cells. The total DNA extracted from the cells was 10 fold serially diluted (from 10.0ng/ µl to 1.0fg/ µl per reaction) and the real-time assay was performed for each dilution (with 5 µl of samples i.e 50.0ng to 5.0fg of DNA) of the CPV genomic DNA in triplicates. The experiments to determine the sensitivity were performed thrice to determine the inter assay variability.
Analytical Specificity of Real-time PCR
Real-time PCR with genomic DNA or cDNA extracted from cell cultures of canine adenovirus 1 and 2 (CAV1 and 2), canine distemper virus (CDV), canine corona virus (CCoV) and canine parainfluenza virus (CPIV) as template to determine the primer specificity. Melt curve analysis were performed to determine non-specific amplification.
Mini-Sequencing
Mini-sequencing reaction as described by Naidu et al. (2012) was performed for all samples that were determined positive in the real-time PCR assay. A modification in the mini-sequencing reaction, where-in only the two pairs of primers viz., CPV Typing 4 S and CPV Typing 5 AS instead of the five pairs of primer sets described in the previous protocol by Nadiu et al. (2012) was used in the assay. Below is the brief description of the protocol The amplicons of the real-time PCR were treated with 5 units of Shrimp Alkaline Phosphatase (SAP; New England Biolabs, Ipswich, USA) and 3 units of Exonuclease I (Exo-I; New England Biolabs, Ipswich, USA) at 37°C for 1 hour, followed by inactivation at 75°C for 15 min. The Exo/SAP treated PCR product was used directly as template in the subsequent mini-sequencing reaction. The mini-sequencing reaction was performed in a total volume of 10µl reaction mixture containing 3µl of Exo I/SAP treated PCR product, 5µl of SNaPshotTM multiplex ready reaction mix (Applied Biosystems, Foster City, USA) and 10 picomoles of each primer (Primers- CPV Typing 4 S, CPV Typing 5 AS;) that differentiate the CPV2 variants CPV2a, 2b & 2c. The reaction conditions were as follows: 28 cycles of denaturation at 96°C for 10s, annealing at 50°C for 5s and extension at 60°C for 30s. The extended products were treated with SAP (1U) followed by heat-inactivation at 95°C for 5 minutes. A 1.0µl of SAP treated product was added with 8.5µl of formamide and 0.5µl of internal size standard GeneScan-120 LIZ (Applied Biosystems, Foster City, USA). The above mixture was then denatured at 95°C for 5 minutes and snap chilled on ice. The samples thus prepared were samples were then processed on an ABI PRISM 3130xl Genetic analyzer. The results were analyzed using the Gene-mapper v.3.7 software (Applied Biosystems, Foster City, USA). PCR amplification products from the standard plasmid constructs were used as positive controls. Similar reactions were also performed with standard plasmid constructs (TOPO-CPV2a, TOPO-CPV2b and TOPO-CPV2c) and the genomic DNA extracted from cell-cultures.
RESULTS
Real-time PCR
Figure 1 presents the amplification plot in the Real-time PCR of TOPO-CPV2b plasmid constructs at various dilutions (1x1010 to 1x10-3). The mean Cq values, standard deviation (SD) and coefficient of variation for each dilution are presented in Table 1. The real-time PCR data for the ten-fold serially diluted CPV type 2b viral genomic DNA (50ng to 5fg per reaction) is also presented in Table 1.
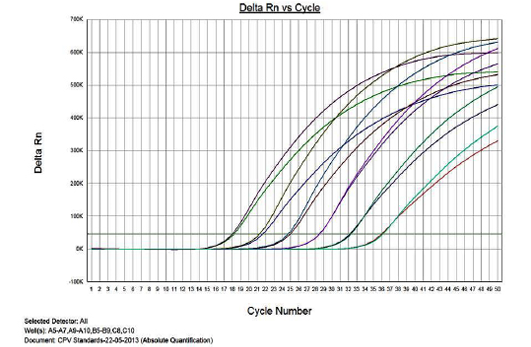
Linearity of the Assay
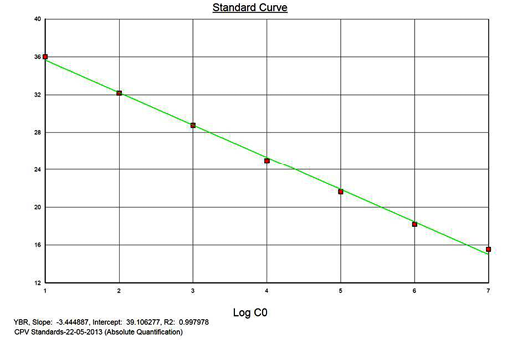
The real-time PCR results for the plasmid construct of CPV2b showed high linearity of detection (r2=0.99) to plasmid copy numbers (Figure 2; Cq vs Copy number plot). The assay was accurate as shown in the low standard errors (SE; Table 1) across triplicates of the copy numbers from log 1 to 7. The dynamic range of detection for the plasmid constructs showed low standard error (SE: 95% confidence interval) for a wide range of dilutions (Table 1). The real-time PCR results with plasmid constructs were hence used to determine the relative concentration of virus in the faecal samples of viral load from the clinical samples (Figure 1). The coefficient of correlation (r2) ranged from 0.98 to 0.99 for the different dilutions of plasmid constructs; the slope ranged from -3.444 to -3.775 and therefore the overall efficiency of the real-time PCR was found to be > 94% (Figure 3). Similar curves of linearity and amplification efficiencies were obtained for the real-time PCRs with plasmid constructs of CPV2a (TOPO-CPV2a) and CPV2c (TOPO-CPV2c).
Similar linearity (r2=0.98±0.01) and amplification efficiency (≥93%; slope= -3.603±0.011) of the assay was also seen with ten-fold serial dilutions of CPV genomic DNA (5ng to 5fg) (Figure 3).
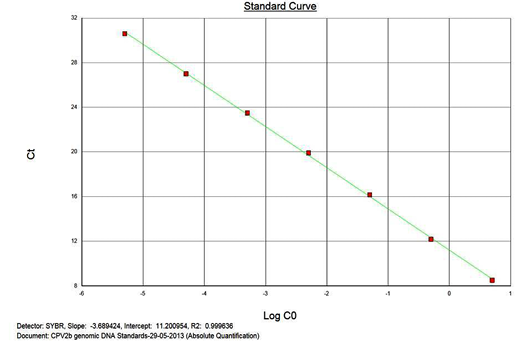
Precision and Variability
To evaluate the inter-assay variability, the assay was performed twice and the coefficient of variation of the assay was determined. The CV values ranged from 0.65 to 3.88 and 0.46 to 3.54 respectively, for plasmid constructs and genomic DNA.
The intra assay variability was determined as coefficient of variation (CV) for both, the plasmid constructs and genomic DNA. The CV was ranged from 0.25 to 1.5 and 0.00 to 1.8 for the plasmid constructs and genomic DNA, respectively (Table 1). The experiment was repeated thrice to determine the precision of the assay. The CV (ranging from 0.41 to 4.27) thus obtained for the intra-assay were well within the accepted range recommended for standards diagnostic test.
Table 2: Screening and typing of CPV field samples with SYBR green real-time PCR followed by mini-sequencing
|
S No |
Sample ID |
Virus Isolation |
HA titer |
PCR |
Real-time PCR |
Copy Number |
Typing by Mini sequencing |
|||
|
Results |
Cq value |
CPV2a |
CPV2b |
CPV2c |
||||||
|
1 |
PCR2.1-CPV2a |
NA |
NA |
+ve |
+ve |
32.21 |
9.90E+01 |
√ |
x |
x |
|
2 |
PCR2.1-CPV2b |
NA |
NA |
+ve |
+ve |
32.4 |
8.90E+01 |
x |
√ |
x |
|
3 |
PCR2.1-CPV2c |
NA |
NA |
+ve |
+ve |
32 |
1.00E+02 |
x |
x |
√ |
|
4 |
PCR2.1-CPV2a+PCR2.1-CPV2b |
NA |
NA |
+ve |
+ve |
32.5 |
8.50E+01 |
√ |
√ |
x |
|
5 |
PCR2.1-CPV2a+PCR2.1-CPV2c |
NA |
NA |
+ve |
+ve |
31.15 |
5.25E+02 |
√ |
x |
√ |
|
6 |
PCR2.1-CPV2b+PCR2.1-CPV2c |
NA |
NA |
+ve |
+ve |
32.3 |
8.91E+02 |
x |
√ |
√ |
|
7 |
CPV-SC-03 |
+ve |
ND |
+ve |
+ve |
23.24 |
9.66E+04 |
√ |
x |
x |
|
8 |
CPV-SC-14 |
+ve |
ND |
+ve |
+ve |
19.51 |
9.16E+05 |
√ |
x |
x |
|
9 |
Hyd-NRG-01 |
ND |
ND |
+ve |
+ve |
25.54 |
1.00E+04 |
√ |
x |
x |
|
10 |
Hyd-NRG-02 |
ND |
ND |
+ve |
+ve |
26 |
1.90E+04 |
√ |
x |
x |
|
11 |
Hyd-NRG-03 |
+ve |
64 |
+ve |
+ve |
16.51 |
5.69E+07 |
√ |
x |
x |
|
12 |
Hyd-NRG-04 |
+ve |
256 |
+ve |
+ve |
15.41 |
1.00E+07 |
√ |
x |
x |
|
13 |
Hyd-NRG-06 |
+ve |
ND |
+ve |
+ve |
18.05 |
1.19E+06 |
√ |
x |
x |
|
14 |
Hyd-NRG-10 |
ND |
ND |
+ve |
+ve |
32 |
1.00E+02 |
√ |
x |
x |
|
15 |
Hyd-NRG-11 |
ND |
ND |
+ve |
+ve |
33.33 |
5.10E+01 |
√ |
x |
x |
|
16 |
Hyd-NRG-12 |
ND |
ND |
+ve |
+ve |
32. 20 |
9.85E+01 |
√ |
x |
x |
|
17 |
Hyd-NRG-13 |
ND |
ND |
ND |
+ve |
ND |
ND |
√ |
x |
x |
|
18 |
Hyd-NRG-14 |
ND |
ND |
ND |
+ve |
38.01 |
1.07E+01 |
√ |
x |
x |
|
19 |
Hyd-NRG-15 |
ND |
ND |
ND |
+ve |
38.8 |
1.00E+01 |
√ |
x |
x |
|
20 |
Hyd-NRG-16 |
ND |
ND |
+ve |
+ve |
33.7 |
3.58E+02 |
√ |
x |
x |
|
21 |
Hyd-NRG-17 |
ND |
ND |
+ve |
+ve |
23.07 |
9.56E+04 |
√ |
x |
x |
|
22 |
Hyd-NRG-18 |
ND |
ND |
ND |
+ve |
40 |
3.00E+00 |
√ |
x |
x |
|
23 |
Hyd-NRG-19 |
ND |
ND |
+ve |
+ve |
31.97 |
1.08E+02 |
√ |
x |
x |
|
24 |
Hyd-NRG-20 |
ND |
ND |
ND |
+ve |
ND |
ND |
√ |
x |
x |
|
25 |
Hyd-NRG-21 |
ND |
ND |
+ve |
+ve |
35.8 |
2.60E+01 |
√ |
x |
x |
|
26 |
Hyd-NRG-22 |
ND |
ND |
ND |
+ve |
ND |
ND |
√ |
x |
x |
|
27 |
Hyd-NRG-23 |
ND |
ND |
ND |
+ve |
39.56 |
2.90E+00 |
√ |
x |
x |
|
28 |
Hyd-NRG-24 |
ND |
ND |
ND |
+ve |
ND |
ND |
√ |
x |
x |
|
29-66 |
Hyd-NRG-25- Hyd-NRG-58 |
ND |
ND |
ND |
ND |
ND |
ND |
NA |
NA |
NA |
*ND- Not detected; NA- Not applicable; +Ve – Positive; √ - typed aginst the same; Serial number 1 to 3 are positive plasmid constructs of CPV2a, 2b and 2c; serial number 4 to 6 are pooled positive plasmid costructs. Serial number 7 to 66 were test samples. The virus genome quantity from the clinical samples were determined with the aid of standard graph which was ranging from 102 to105 copies in 50ul of sample
Sensitivity (SE) and Specificity (SP)
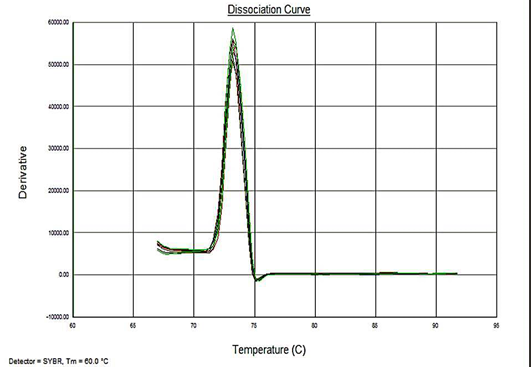
Sensitivity of the assay was determined as lower limit of detection (LOD). For the plasmid constructs LOD was 1x100 copies and 5fg for the genomic DNA. Melt curve analyses were performed to determine the specificity of the assay. The melting temperature for the amplification product of the target DNA (VP2 -145 bp fragment) was ~73.3°C (Figure 4). The analysis showed no evidence of amplification of non-specific DNA or the formation of Primer dimers contributing to the measured fluorescence.
The specificity of the primer pairs to the target DNA was evaluated in-silico in the online tool-http://insilico.ehu.es/PCR/ and also in the real-time PCR assays with. Genome from closely related viral genera viz., canine adenovirus 1 and 2 (CAV1 and 2), canine distemper virus (CDV), canine corona virus (CCoV), canine parainfluenza virus (CPIV). No amplifications were seen either in-silico or relative PCR assays.
Typing by Mini-sequencing
The mini-sequencing technique (Naidu et al., 2012) of the present study could differentiate CPV2a, CPV2b and CPV2c. When the respective primers were used, even plasmid constructs at as low as single copy (Figure 5) and for genomic DNA at the lowest concentration of 5.0 fg/reaction could be detected.
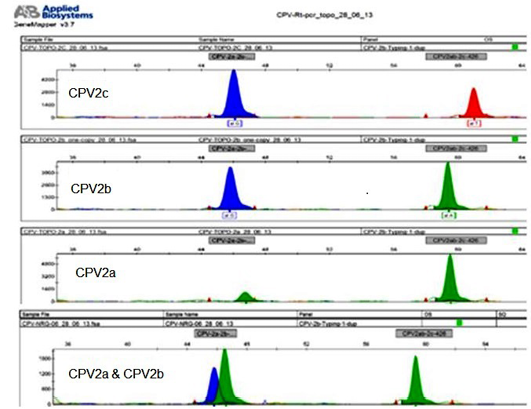
Figure 5: The sample that are detected with low plasmid copy number 1x100 per 5ul (CPV2b) and genomic DNA cocentarion of 5fg per assay, and differentiation between CPV variants
Screening Field Samples
A total of 60 faecal samples from dogs showing enteric symptoms14 samples were positive by PCR (Mochizuki et al., 1993b; Senda et al., 1995; Pereira et al., 2000). Twenty two samples were found positive by SYBR green real-time PCR assay and 5, 2 samples were positive by virus isolation and HA test respectively. The mini-sequencing results showed that the variant type of the virus in all the 22 samples determined positive by real-time PCR assay was CPV type 2a (Table 2).
DISCUSSION
Published literature on the infection epidemiology suggests that the variant strains of CPV (CPV2a, 2b, 2c) have completely replaced the original CPV2 genotype world-wide, (Parish et al., 1985, 1991; Buonavoglia et al., 2001; Nakamura et al., 2004; Decaro et al., 2005a). Though real-time PCR based CPV detection methods are high in sensitivity (Decaro et al., 2005c; Lijun Shi et al., 2012; Chao-Nan Lin et al., 2014) it does not determine the variant types involved in the infection. This since the variants differ by a single amino acid from each other in their VP2 sequence resultant of a corresponding DNA sequence variation was in single nucleotide at either the second or third base of the codon. Hence it was reasoned that a method for identification of single nucleotide polymorphism (SNP) is more appropriate for identifying CPV2 variant types (Naidu et al., 2012; Chinchkar et al., 2006). A real-time PCR assay using MGB probes have been published for the detection and differentiation of the CPV2 variant types. A significant limitation of this assay, apart from relatively high cost of MGB probes is that it is time consuming (involves setting up of multiple reactions for strain specific identification of the virus, (Deacro et al., 2005b, 2006b). A Haemagglutination assay using type specific monoclonal antibody was also used for CPV diagnosis and typing but suffers from the specificity and sensitivity issues (Decaro et al., 2005b). Our approach here has been to develop an economical, sensitive yet specific assay for the diagnosis of CPV infection as well as identification of CPV2. In the present study, SYBR green real-time PCR was combined with the mini-sequencing based typing technique (Naidu et al., 2012) for detection and type specific differentiation of CPV infection. The current approach allowed rapid detections, quantification and typing of CPV directly from faecal samples.
The SYBR green real-time assay was targeted at the 145 bp region of VP2 gene that harbors type specific SNPs such that the amplification product of the real-time assay serves as the template for typing by the mini-sequencing technique. The real-time PCR was highly sensitive (LOD of plasmid constructs = 1 copy; and genomic DNA= 5fg) and found to be in agreement with other published reports (Decaro et al., 2005c; Decaro et al., 2006a, 2006b, 2006c; Lijun Shi et al., 2012; Chao-Nan Lin et al., 2014). The assay also qualifies the recommended statistical criterion of low SE (0.051). The assay may thus be considered for extensive application in the detection of CPV directly from the faecal samples.
The mini-sequencing technique could type virus from as little as a single copy of virus specific DNA (based on plasmid copy number). The concentration of virus genome in faecal samples (faecal samples) ranged from 10 to 60 copies as determined from the real-time PCR assay with plasmid constructs as standard. Hence the mini-sequencing technique is seems to be sensitive enough for typing directly from clarified faecal samples; and do not require extensive processing or concentration of virus. The sensitivity of the mini-sequencing reaction technique in the present study is found to be higher than the previous study by Naidu et al. (2012). This is because of the smaller size (145 bp) of the target region when compared to much longer target of previous report (576 bp). The sensitivity of the mini-sequencing based typing of variants is high and in the current study all the positive samples could be unambiguously typed as CPV2a variant. Even though in the present study, all the sample are typed as CPV2a, but the principle of mini-sequencing technique reported earlier and adopted here could differentiate all CPV variants effectively. The same was demonstrated using the plasmid construct harboring the VP2 fragment of different variants.
The methodology of the combined technique of real-time PCR and mini-sequencing supports high-through put detection of the field infection as well as determination of variant types involved in the infection. Besides, the technique is rapid and involves limited processing of faecal samples. Hence this combined approach may have the potential for extensive application in epidemiological studies.
CONCLUSION
Detection of CPV infection by the SYBR green based real-time PCR assay and differentiation of the virus type by mini-sequencing technique from the amplified PCR product is a rapid, sensitive and specific approach. The protocol can be implemented extensively for diagnosis of CPV and differentiation of CPV types in epidemiological studies.
AUTHOR’S CONTRIBUTION
Hari Prasad Naidu G carried out laboratory work, conducted the experiments, isolated the viruses used in the present study, analysed the data and prepared the manuscript. Surendra KSNL helped in designing the laboratory work and analysed the data. Rana SK designed the study, facilitated the study by arranging samples and finalised the manuscript. Subramanian BM provided expert opinions on the design of work and assisted in analysis. Ponnanna NM helped in reviewing the data and manuscript. Sharma GK helped in facilitating the study, provided the administrative support and facilitated financial support from the NDDB. All the authors are thankful to Srinivasan VA for conceptualisation of the work from time to time, Hw also critically reviewed the manuscript and provided a meaningful conclusion from the data generated from the present study.
ACKNOWLEDGEMENTS
The authors are grateful to the management of the National Diary Development Board (NDDB) Anand, for providing the facilities to carry out this work, conducted at the Research and Development Laboratory, NDDB, Hyderabad. The author G. Hariprasad Naidu expresses his gratitude to the Indian Immunologicals Limited, Hyderabad, for accord the necessary permission for persuing the doctoral course.
CONFLICT OF INTEREST
These exists no conflict of interest.
REFERENCES

