
Advances in Animal and Veterinary Sciences


Review Article
Advances in Animal and Veterinary Sciences 2 (4S): 55 – 63Special Issue–4 (2014) (Reviews on Frontiers in Animal and Veterinary Sciences)
New Approaches for Diagnosis of Viral Diseases in Animals
Prasad Minakshi1*, Koushlesh Ranjan2, Basanti Brar1, Supriya Ambawat1, Mukhtar Shafiq1, Alisha Alisha1, Pawan Kumar1, Joshi Vinay Ganesharao1, Savi Jakhar1, Shweta Balodi1, Anjali Singh1, Gaya Prasad1
- Department of Animal Biotechnology, LLR university of Veterinary and Animal Sciences, Hisar, Haryana, India, 125004
- Department of Veterinary Physiology and Biochemistry, Sardar Vallabhbhai Patel University of Agriculture and Technology, Meerut, Uttar Pradesh, India, 250110
*Corresponding author:[email protected]
ARTICLE CITATION:
Minakshi P, Ranjan K, Brar B, Ambawat S, Shafiq M, Alisha A, Kumar P, Ganesharao JV, Jakhar S, Balodi S, Singh A, Prasad G (2014). New approaches for diagnosis of viral diseases in animals. Adv. Anim. Vet. Sci. 2 (4S): 55 – 63.
Received: 2014–03–10, Revised: 2014–05–11, Accepted: 2014–05–13
The electronic version of this article is the complete one and can be found online at
(
http://dx.doi.org/10.14737/journal.aavs/2014/2.4s.55.63
)
which permits unrestricted use, distribution, and reproduction in any medium, provided the original work is properly cited
ABSTRACT
Identification and control of viruses that causes diseases in human, domestic and wild animals represent continuous challenges to medical and veterinary sciences. From centuries many diagnostic techniques have been tried to identify individual infectious agents by their clinical signs and symptoms produced in the susceptible host. But it is not always possible to identify or differentiate the disease on the basis of its signs and symptoms and thus requires other advanced diagnostic procedures to clearly identify and confirm the pathogens. Rapid detection of infectious agents in animals as well as in environment is essential for effective control of infectious diseases. There are various approaches for detection of viral diseases which includes conventional as well as modern approaches. Several conventional techniques such as isolation in cell culture, serology and histological identification are traditionally used to identify viral pathogens. However, the latest molecular diagnostic technologies are more advantageous as they are offer more sensitive, less time consuming, high throughput result. Other novel technologies such as polymerase chain reaction (PCR), Reverse Transcriptase PCR (RT–PCR), Real Time–PCR, DNA probe, nucleic acid sequencing etc. provide a thorough understanding of accurate diagnosis and discrimination of present and emerging diseases. The aim of these new tools is to detect the presence of pathogen before the onset of disease. Here we have discussed the advancement of diagnostic tools and their applications for important animal viruses of domestic and wild animals.
INTRODUCTION
Viruses are serious threats to human and animal health. Only few successful antiviral treatments are available for viral infections. Therefore, early and rapid detection and characterization of viral pathogens are essential. The viral disease diagnosis is important for not only deciding the treatment strategies but also to identify the prevalence of viruses in different forms such as serotypes or isolates. Many viral diseases like bluetongue virus, peste des petets virus, classical swine fever virus, Newcastle disease virus of poultry, foot and mouth disease virus have continuously raising serious economic problems in the field. Bluetongue virus (BTV) has been in the forefront of molecular virology and is one of the best understood viruses at the molecular and structural level (Roy, 2008). For bluetongue virus (BTV), specific peptides representing its antigenic epitopes and ssRNA–binding domains have been utilized to develop unique diagnostics that can differentiate whether an animal is vaccinated with BTV vaccine or infected by BTVs. The gene–silencing tool has been used for RNA inference (RNAi) for determination of gene functions and as potential therapeutics for Bluetongue disease. The signature amino acid sequences have been identified to develop a peptide–based assay to differentiate a BTV–vaccinated animal from an animal infected with BTV. Other viruses such as Rotavirus (Niture et al., 2008; Minakshi et al., 2004), Parvovirus (Savi et al., 2009), Caliciviruses (Yu et al., 2013), Paramyxoviruses (Killip et al., 2012) and Influenza virus (Tonnessen et al., 2013) have also been extensively studied at the molecular level. Various molecular techniques such as polymerase chain reaction (PCR), probe hybridization (Minakshi et al., 2005), microarray, nucleic acid sequencing etc. have been extensively used for this purpose. In this paper molecular techniques for detection of viruses are reviewed and the potential for their application are discussed. The application of new techniques as a routine tool in a diagnostic laboratory is an area where relevant literature is not available and this may contribute to the acceptance of these methods.
Conventional Approaches
The viral disease diagnosis includes the isolation of live virus in different cell cultures, embryonated eggs or through animal inoculation. The detection of viral proteins (antigens) and viral genome fragments (viral nucleic acid) are also carried out. In general, various viral disease diagnostic tests can be grouped into 3 broad categories:
Direct Examination of Specimen
Direct examination methods are often used for rapid diagnosis of viral diseases. This is highly useful in clinical management of diseases especially when treatment depends on the rapid availability of laboratory results.
Antigen Detection
Virus isolation is the most effective and standard method for viral disease diagnosis. Growth of viruses was initially restricted to animal inoculation. For BTV diagnosis initially sheep inoculation was used as the diagnostic method. Subsequently developing chicken embryo was used for isolation of the virus. Now a day, cell culture is used for production of large quantities of live viruses (e.g. BTV in BHK–21 or Vero cell line) to produce diagnostic antigens (Figure 1).
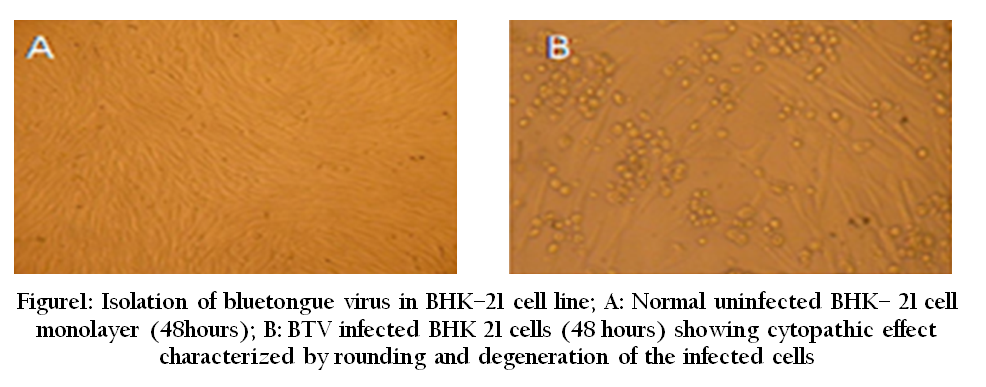
Microscopic Examination
The live replicating viruses often produce cytopathic changes in infected cells which can be easily observed by light microscopy. However, low sensitivity is the biggest limitation of this method as a diagnostic tool. In electron microscopy, specimens are stained negatively with phosphor–tungstate, or sometimes uranyl acetate and then scanned by electron microscopy (Figure 2). The specific modification in the basic EM was made by using immune–gold electron microscopy by adding specific antibodies labeled with gold (Prasad et al., 1993).
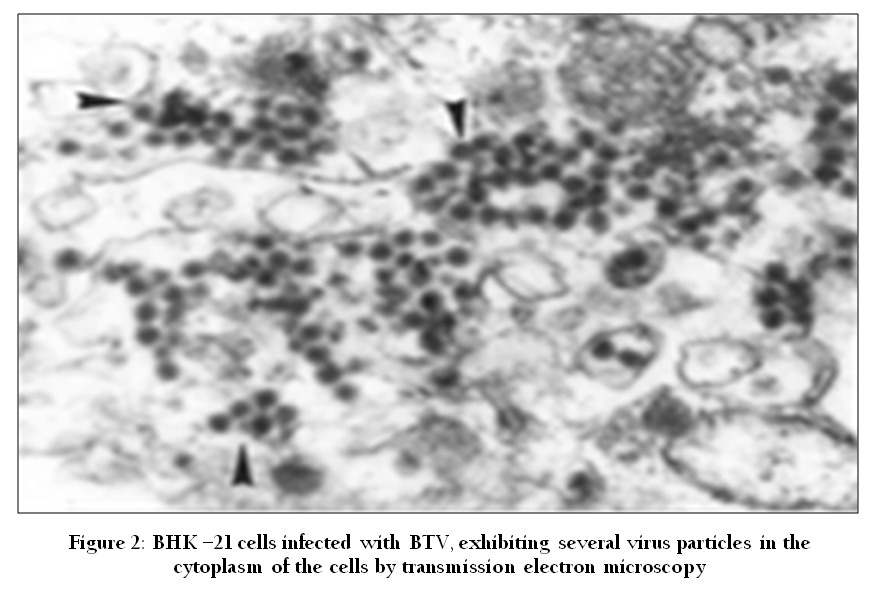
Figure 2: BHK –21 cells infected with BTV, exhibiting several virus particles in the cytoplasm of the cells by transmission electron microscopy
Indirect Examination/Virus Isolation
Virus isolation is one of the best methods for viruses that can be grown in vitro in cell culture, such as FMD virus (FMDV), Newcastle disease virus, Avian influenza virus (AIV) etc. Although it is time consuming, but this method is very sensitive, and offers the maximum opportunity for subsequent viral characterization. Cell cultures, chicken embryo and animal inoculation may be used for virus isolation. Various cell lines have been established for viral isolation such as N2A for rabies virus, procine kidney cell line for classical swine fever virus, Vero for NDV and PPRV, BHK–21 for BTV, MA104 for rotaviruses, MDCK for canine parvovirus. Viruses can be adapted in different cell lines which include primary (Monkey Kidney), semi–continuous cells (Human embryonic kidney) and continuous cultures (HeLa, Vero cell line). Confirmation of the presence of virus in cell culture may be carried out using neutralization, immunofluorescence or molecular tests (Mishra et al., 2002; Prasad and Minakshi, 1999) (Table 1).
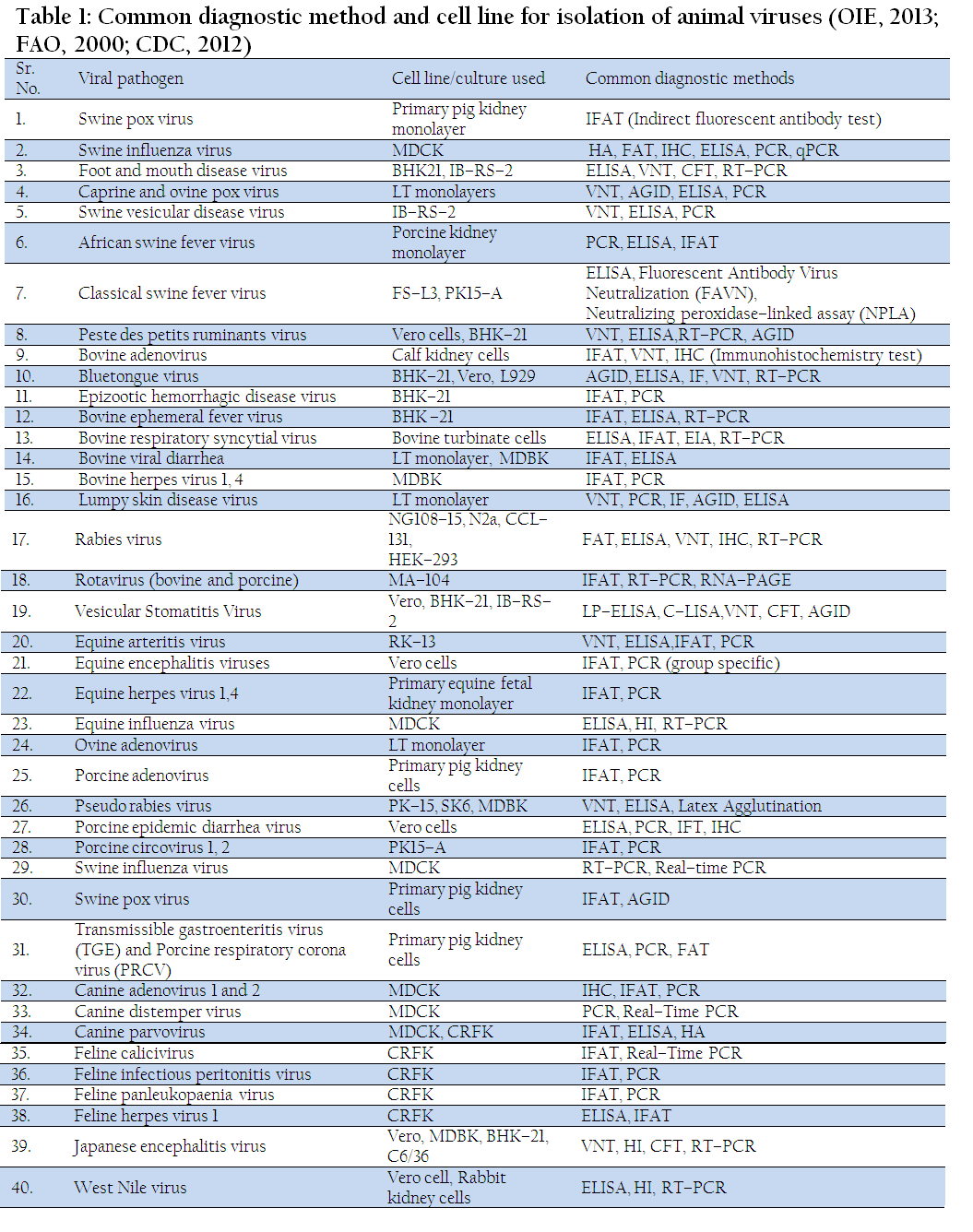
Table 1: Common diagnostic method and cell line for isolation of animal viruses (OIE, 2013; FAO, 2000; CDC, 2012)
Serological Examinations
Several types of serological tests are available for diagnosis of viral antigen or antibodies. Some of the assays such as enzyme immuno assay (EIA) and radio immuno assay (RIA) are used specifically for IgM or IgG, whereas other assays such as compliment fixation test (CFT) and Haemagglutination Inhibition (HAI) can only detect total antibody comprising mainly IgG. Newer techniques such as EIAs offer better sensitivity, specificity and reproducibility than classical techniques such as CFT and HAI. Certain viruses such as influenza, parainfluenza, flaviviruses etc. have been detected using Haemagglutination Inhibition (HAI) test. TheAgar gel immunodiffusion (AGID) test and dot immunobinding assays were employed for canine distemper virus (CDV) and feline parvovirus (FPV) specific antibody detection in Asiatic lions.
Enzyme Linked Immunosorbent Assay (ELISA)
ELISA is now the most widely used form of immunoassay for the rapid detection of viral antigens and antibodies. There are many different formats of ELISA are available such as DOT blot (Figure 3) and Plate method. DOT blot method has been used for Rotavirus, Bovine herpes virus and BTV detections. Commercial ELISA kits were also used for diagnosis of several viral diseases such as feline immunodeficiency virus (FIV) and feline leukemia virus (FeLV) (Ramanathan et al., 2007). The antibodies against bluetongue virus in deer, camel and sheep have been detected using AGID (Naresh et al., 1996). The bluetongue specific antibody in domestic and wild ruminants in and around Sariska tiger reserve in Rajasthan has been detected in deer as well as sheep (Prasad et al., 1998). Peptide based ELISA are also available for diagnosis of various viruses like infectious bursal disease virus, PPRV, FMDV, SARS and Infectious bronchitis (Dechamma et al., 2006, Jackwood et al and Hilt, 1995, Joshi et al., 2013a).
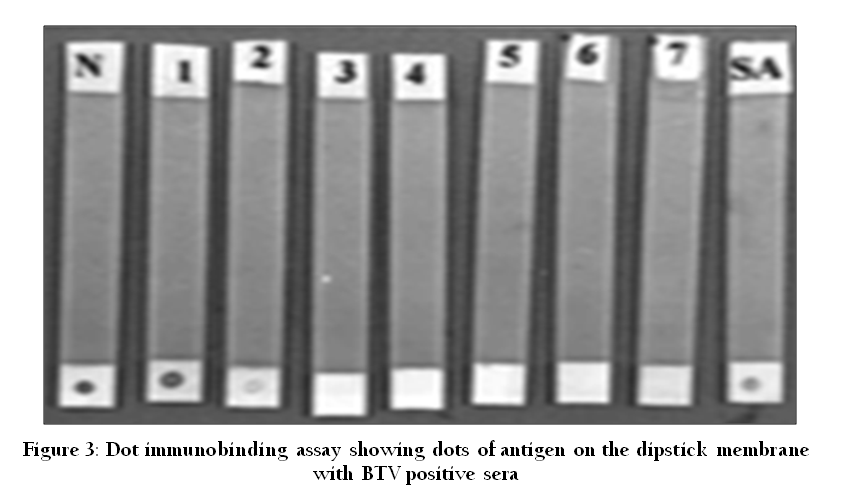
Figure 3: Dot immunobinding assay showing dots of antigen on the dipstick membrane with BTV positive sera
Immunofluorescense
Immunofluorescence (IF) assay is commonly used for the rapid detection of virus infections by the detection of virus specific antigen in clinical samples, as well as the virus–specific antibody. The technique is primarily based on reaction between fluorescein– labeled antibody and virus specific antigens in specimens (cells). The cells containing virus specific antigen fluoresce under UV illumination (Figure 4). The virus specific antibody can be detected using indirect IF whereas virus specific antigen is detected using direct or indirect IF. The in–vitro susceptibility of goat mononuclear cells to bluetongue virus has been studied in Haryana using IF test (Garg et al., 1996).
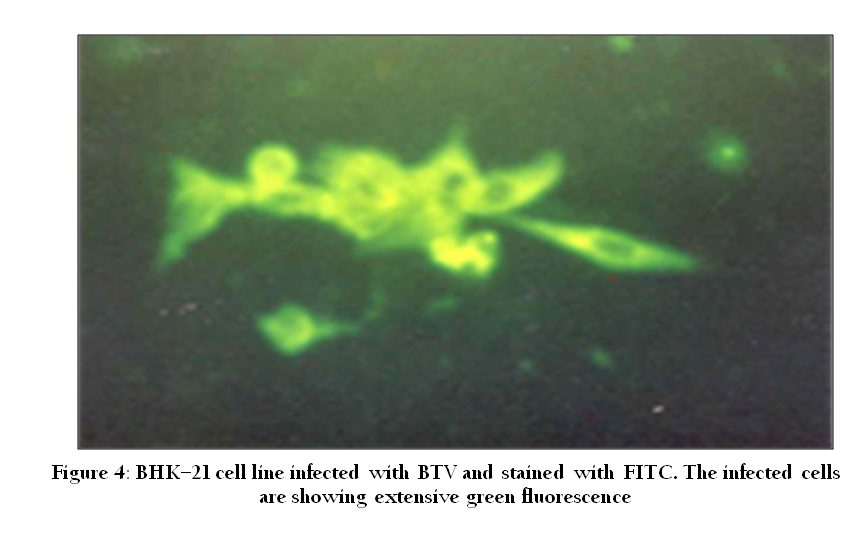
Figure 4: BHK–21 cell line infected with BTV and stained with FITC. The infected cells are showing extensive green fluorescence
Molecular Approaches
Various molecular biology techniques have been used for direct detection of viral genome (nucleic acid) in the specimen which plays an important role in clinical virology. In the molecular techniques, DNA is extracted from the sample of interest which can be probed by DNA hybridization and analyzed by restriction fragment length polymorphism (RFLP). More commonly, DNA is amplified by the polymerase chain reaction (PCR) using pathogen specific primers for diagnosis. The amplified PCR products can be sequenced for confirmatory diagnosis. A number of assays like RNA–polyacrylamide gel electrophoresis (RNA–PAGE), reverse transcription polymerase chain reaction (RT–PCR) etc have been employed for Newcastle disease virus, rotavirus, parvovirus and bluetongue virus diagnosis (Dalal et al., 2008; Kovi et al., 2007; Dahiya et al., 2005, Mase and Kenehira, 2012).
Nucleic Acid Probes
Nucleic acid probes are segments of DNA or RNA that have been labeled with enzymes, antigenic substrates, chemiluminescent moieties or radioisotopes. They are highly specific to pathogen specific nucleic acid. Probes can be directed to either DNA or RNA targets and can be from 20 to thousands of bases long. The presence and the quantity of hybrids are determined by the detection of the labeling of probe. A variety of labeling and detection systems exist for nucleic acid probes such as radiolabelled (Figure 5) and non–radiolabelled methods. These include labeling with a variety of haptens such as biotin or digoxygenin (DIG) and detection by antibody binding coupled with fluorescent, chemiluminescent or colorimetric detection methods (Minakshi et al., 2005). Recently new category of nucleic acid probe called peptide nucleic acids are also used for molecular diagnosis of viral diseases (Joshi et al., 2013b).
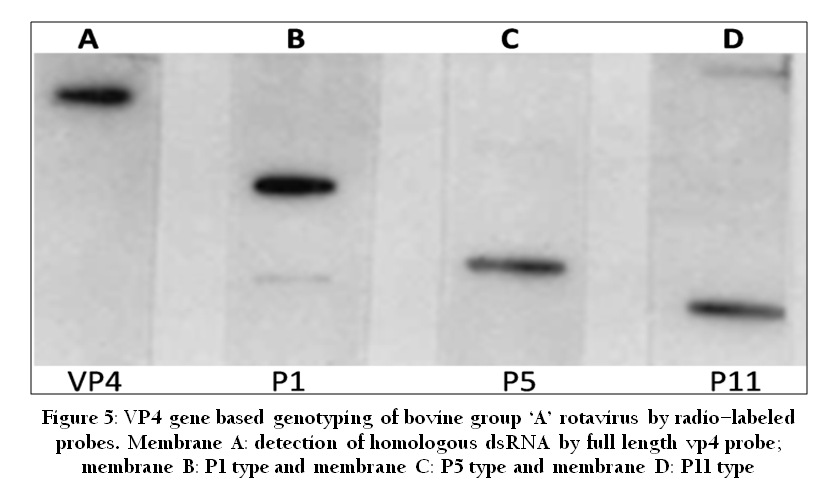
Figure 5: VP4 gene based genotyping of bovine group ‘A’ rotavirus by radio–labeled probes. Membrane A: detection of homologous dsRNA by full length vp4 probe; membrane B: P1 type and membrane C: P5 type and membrane D: P11 type.
Polymerase Chain Reaction (PCR)
Polymerase chain reaction is a technique for in–vitro amplifying a specific region of DNA by a thermo stable DNA polymerase (Figure 6). PCR reaction uses short oligonucleotide primer sequences, dNTPs, DNA template and thermo stable DNA polymerase enzyme. PCR reaction involves three steps viz. denaturation, annealing and extension repeatedly for 30–40 cycles in thermal cycler. It can be used for detection of virus specific nucleic acid. There are several modifications of PCR such as RT–PCR, Real–Time PCR, Multiplex PCR and Multiplex real–time PCR etc. used in molecular diagnosis of viruses. The PCR amplified products can be used for nucleic acid sequencing to confirmatory diagnosis and detection of phylogenetic variations (Figure 7) and new strains of pathogens (Susmitha et al., 2012).
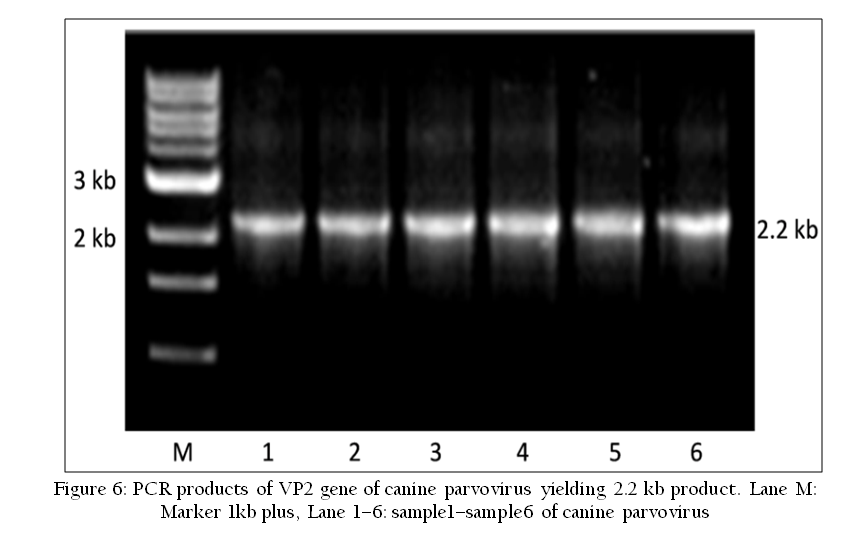
Figure 6: PCR products of VP2 gene of canine parvovirus yielding 2.2 kb product. Lane M: Marker 1kb plus, Lane 1–6: sample1–sample6 of canine parvovirus.
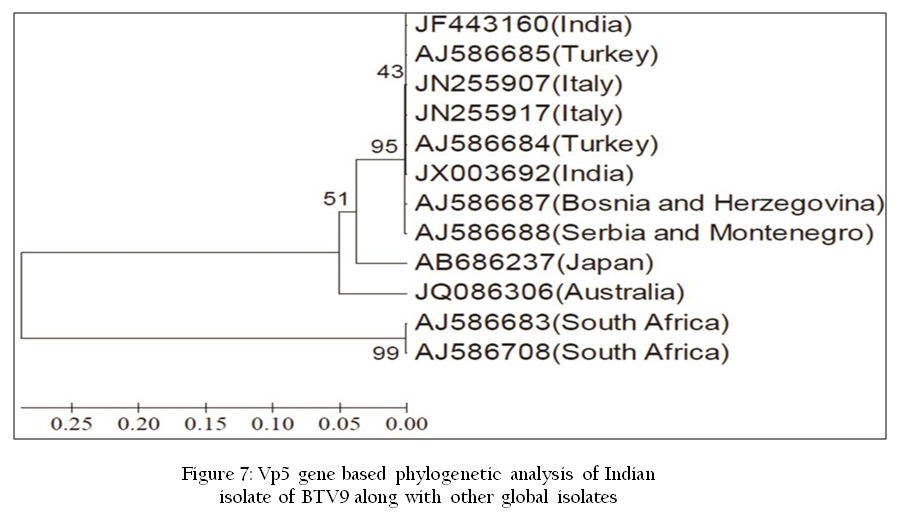
Figure 7: Vp5 gene based phylogenetic analysis of Indian isolate of BTV9 along with other global isolates
Reverse Transcription Polymerase Chain Reaction (RT–PCR)
In RT–PCR, the RNA template is first converted into a complementary DNA (cDNA) using a reverse transcriptase enzyme (eg. Moloney murine leukemia virus enzyme). The cDNA is then used as a template for exponential amplification using pathogen specific oligonucleotide primer in PCR. The RT–PCR is currently the most sensitive method of RNA detection available. In molecular virology RT–PCR is used for diagnosis of RNA viruses such as Rotavirus and BTV. The sensitivity and specificity achieved in a well–designed RT–PCR make it an ideal tool for use in the surveillance and monitoring of many viral infections. It has been used to detect and identify the specific G and P genotypes present in rotavirus (Harimohan, et al., 2012). Similarly, segment 2 and segment 6 based RT–PCR has been standardized for BTV diagnosis (Kumar et al., 2013; Ranjan et al., 2013).
Poly Acrylamide Gel Electrophoresis (PAGE) Analysis
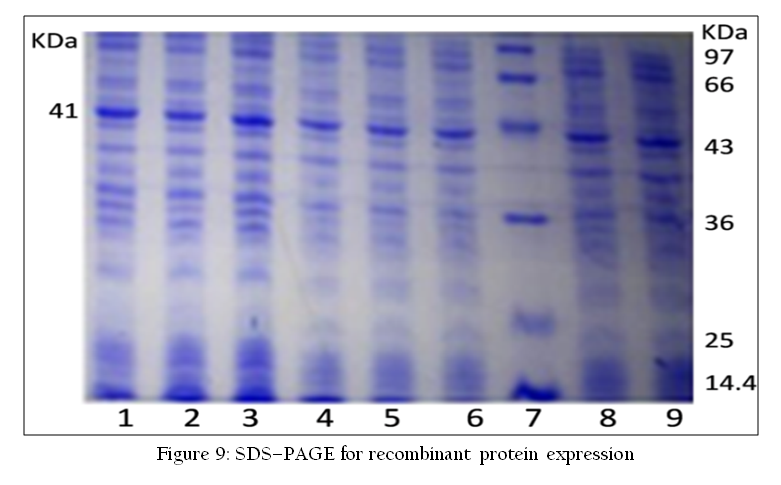
The RNA–PAGE has been used as a confirmatory tool for the identification of segmented genome dsRNA viruses such as Rota virus and bluetongue virus. Viral dsRNA are extracted either from cell culture grown samples or directly from various biological samples such as blood, semen or vector. The dsRNA can be analyzed by 8% RNA–PAGE followed by silver staining using the discontinuous buffer system without SDS (Laemmli, 1970; Svensson et al., 1986). Recently, a highly sensitive silver staining method for BTV has been developed (Minakshi et al., 2013).The characteristic genomes of Rota virus or BTV are visualized after silver staining (Minakshi et al., 2011a) (Figure 8). RNA–PAGE is a sensitive test and can detect up to 105 TCID50/ml of BTV genomic segments (Prasad and Minakshi, 1999). RNA–PAGE can be used for electropherotyping in rotavirus (Harimohan et al., 2012) where several distinct RNA patterns can be observed (Figure 8) while SDS–PAGE can be used for visualization of recombinant protein expression (Figure 9).
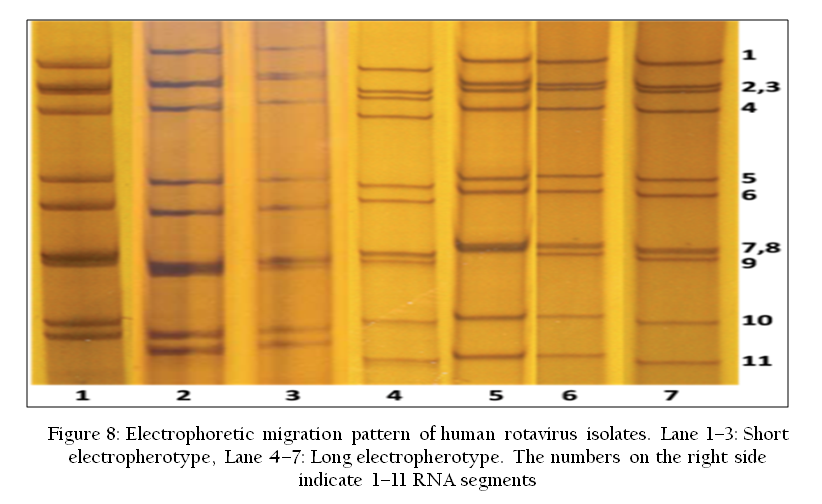
Figure 8: Electrophoretic migration pattern of human rotavirus isolates. Lane 1–3: Short electropherotype, Lane 4–7: Long electropherotype. The numbers on the right side indicate 1–11 RNA segments;
Real–Time PCR
A more recent development in PCR is measurement of concentration of PCR products in real–time as the reaction proceeds PCR. It enables the viral load in a sample to be measured. Real–time PCR uses a beam of light of a specified wavelength with the capacity to illuminate sample in a thermal cycler with a capacity to detect the fluorescence emitted by the excited fluorophore. Real–time RT–PCR is a highly sensitive method that can be used for the laboratory detection of viral RNA from a variety of biological samples such as blood, tissue etc. However, in real–time RT–PCR reaction, viral RNA is reverse transcribed into cDNA and subsequently amplified and detected. The primers and probe combination for each real–time RT–PCR reaction are specific and designed to amplify and detect a targeted region of the viral genome (Vishwaradhya et al., 2013).
Multiplex PCR
In conventional PCR single set of primer is used at a time. However in multiplex PCR, several primer sets are used to allow amplification of multiple templates within a single reaction. It has been used for diagnosis of bluetongue virus, feline herpes virus, porcine epidemic diarrhea virus and transmissible gastroenteritis virus. Multiplex PCR enables the presence of nucleic acids from several pathogens to be detected in single reaction. However, care must be taken to avoid interference between different primer pairs or templates. It is also time and cost–efficient method and has been successfully used in several areas of nucleic acid diagnostics such as gene deletion analysis (Satish et al., 2011).The multiplex PCR was developed for molecular diagnosis of Indian FMDV serotypes O, A, Asia1 and C has demonstrated 100% efficiency in FMDV diagnosis with better diagnostic efficiency than ELISA (Giridharan et al., 2005). Recently, segment 6 based multiplex PCR for simultaneous detection of Indian isolates of BTV serotype 1, 2, 10, 16 and 23 has been standardized (Surendar and Reddy, 2012).
Multiplex Real–Time PCR
The multiplex real–time PCR utilizes two to four fluorogenic oligoprobes for the discrimination of multiple amplicon. So far only a few truly multiplexed real–time PCR assays have been validated successfully. The use of non–fluorescent quenchers and the continuous development of better light sources in the machines are now in use. Now this technique is in use for detection of viral diseases (Read et al., 2011).
Restriction Fragment Length Polymorphism (RFLP)
The RFLP technique differentiates pathogens by analysis of restriction patterns derived from cleavage of their nucleic acid using specific restriction endonuclease enzyme. Isolation of sufficient DNA for RFLP analysis is time–consuming and labor intensive. However, PCR can be used to amplify very small amounts of DNA, usually in 2–3 hours to the levels required for RFLP analysis. Thus, more samples can be analyzed in a shorter time. RFLP was used to differentiate the Canine Parvovirus2 (CPV–2) antigenic variants (Savi et al., 2009) (Figure 10).
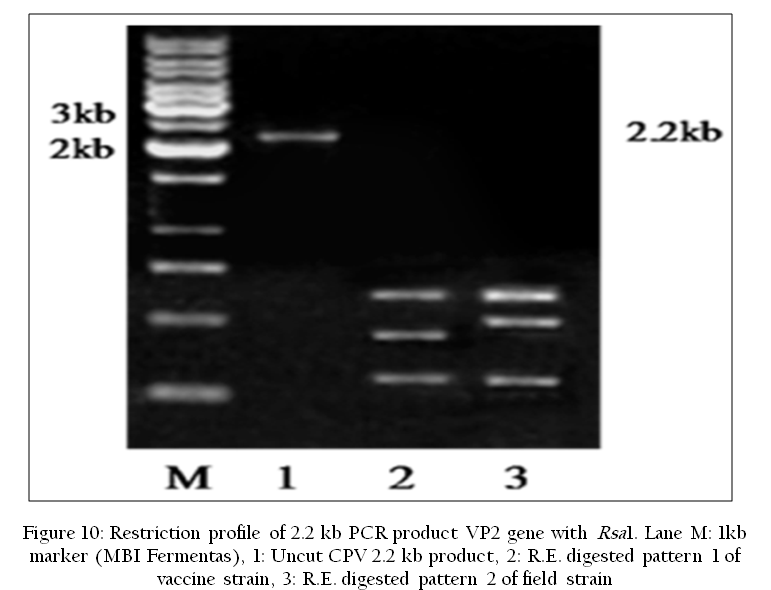
Figure 10: Restriction profile of 2.2 kb PCR product VP2 gene with Rsa1. Lane M: 1kb marker (MBI Fermentas), 1: Uncut CPV 2.2 kb product, 2: R.E. digested pattern 1 of vaccine strain, 3: R.E. digested pattern 2 of field strain
DNA Microarrays
DNA microarrays can be used for the detection of unique DNA (or RNA) sequences. Thus it can be useful in the diseases diagnosis. Microarrays showed advantages over other traditional methods in automation in process, high sensitivity, rapid detection and low cost. In this technique the known DNA is immobilized on small solid supports such as glass slides, nylon membranes or silicon chips. The unknown DNA in the liquid phase is labeled to make probe. DNA or RNA is extracted from various biological samples, and subjected to RT–PCR and/or PCR amplification and labeled with a fluorescent dye. This labeled DNA is then hybridized with the microarray. The specific patterns of fluorescence are detected by a microarray reader which allows the identification of specific gene sequences. DNA microarrays have a potential to diagnose multiple viral pathogens including BTV (Jack et al., 2009).
Nucleic Acid Sequencing
Nucleic acid sequencing has provided an opportunity to study viral epidemiology by providing information about geographical origin of viral isolate. The determination of the nucleic acid sequences may provide information on where the virus came from. The application of nucleic acid sequencing is beyond just diagnosis and characterization of viruses. For example, VP2 and VP5 proteins of BTV are highly variable and determine the serotype specificity (Kumar et al., 2013; Ranjan et al., 2013). The nucleotide sequence and the phylogenetic analysis of these genes can provide a rapid approach for characterization of circulating BTV strains in particular geographical region.
Although, individual gene sequencing of viruses of veterinary importance is quite common, however, the continuing evolution of virus under selection pressure have forced researcher to go for full genome sequencing. Similarly to identify reassortment in viruses having segmented genome, complete genome sequence becomes essential. The full genome sequence of BTV isolates makes it possible to track any changes or reassortment events that occur in the virus isolates. In India full genome sequencing has been carried out for BTV serotype 16 (Minakshi et al., 2012).
NUCLEIC ACID SEQUENCING METHODS
First Generation Sequencing
First generation sequencing technologies include the Maxam–Gilbert method, discovered by Allan M. Maxam and Walter Gilbert (chemical method), and the Sanger method (or dideoxy method), discovered by Frederick Sanger. However, the Sanger method was more commonly used. Fundamental to this method is use of di–deoxy nucleotides, which lack a 3` hydroxyl group, along with the normal deoxy nucleotides. Here DNA chains are synthesized on a template strand. However, when one of the four possible di deoxy nucleotides added, the chain growth is stopped and prevents the further addition of coming nucleotides. In this way a population of nested, truncated DNA molecules are produced that represent each of the sites of that particular nucleotide in the template DNA. The truncated DNA molecules are the allowed for electrophoresis and DNA molecules are separated according to size on an electrophoresis gel. Thus, correct nucleotide sequence is deduced. In 1986, the laboratory of Leroy Hood at Caltech, in collaboration with Applied Biosystems (ABI), developed the first automatic DNA sequencer (Smith et al., 1986) which is now commonly used. The segment 6 and 3 of Indian isolates of BTV has been sequenced using ABI 3130xl genetic analyser (Ranjan et al., 2012; Minakshi et al., 2011b).
Second or Next Generation methods
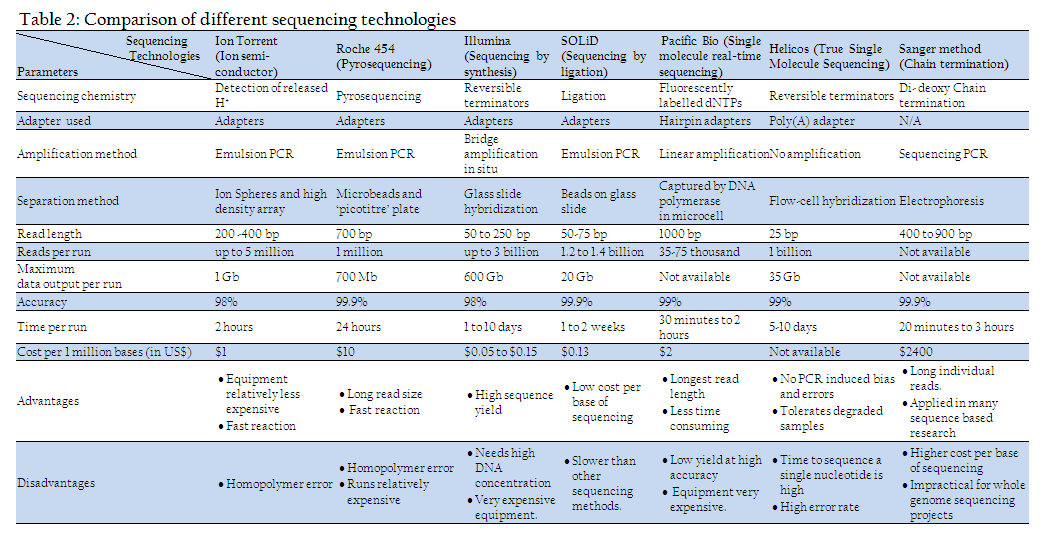
Although first generation sequencing machines are commonly used, but they are of low through put type. To sequence whole genome second or next generation machines are developed. They are high through put sequencing methods enable many DNA fragments (sometimes on the order of millions of fragments) to be sequenced at one time and are much faster than first–generation machines. Four platforms for next generation sequencing are currently in worldwide use: the Roche/454 FLX, the Illumina/Solexa Genome Analyzer, Ion Torrent/Personal Genome Machine (PGM) and the Applied Biosystems SOLiDTM System. Recently, another two massively parallel systems were introduced: the Helicos, HeliscopeTM and Pacific Biosciences, SMRT instruments. These platforms are quite diverse in sequencing biochemistry array generation, read length generated, maximum data output per run, cost per base of sequencing etc (Table 2). However, their work flows are similar. The sequencing process starts with library preparation which is done by random fragmentation of DNA molecule, followed by in–vitro ligation of adaptor sequences to fragmented DNA molecules. The library sequences are then amplified through many different approaches such as emulsion PCR or bridge PCR depending upon type of machine used. The full genome sequence of one of the Indian isolate of BTV serotype 16 has been done using Ion Torrent –PGM (Minakshi et al., 2012).
CONCLUSION
Isolation of pathogen from various field samples will remain as gold standard for viral disease diagnosis. However, rapid detection and molecular characterization of the viral agent in clinical samples on the basis of nucleic acid sequence are very powerful tools for clinical and epidemiological studies. Other modern tests such as micro array, real–time PCR, next generation sequencing etc. are demand of modern era for rapid and reliable diagnosis. Moreover, very less is known about the molecular basis of pathogenesis and virulence of viral pathogens. There are still many situations where live virus in cell culture or animal hosts is inoculated for study of pathogenesis and virulence of virus. In addition, the speed and multiplexing capacity of PCR–based technologies have limitations not to detect live virus. While nucleic acid based diagnostic tests will have clear significance as rapid tests, virus isolation will remain an important component of viral disease diagnosis of domestic as well as wild animals.
ACKNOWLEDGMENT
The study reported in present paper was funded by Indian council of Agricultural Research and Dept. of Biotechnology, New Delhi. The authors are thankful to Dept. of Animal Biotechnology, LLR University of Veterinary and Animal Sciences, Hisar for providing infrastructural facility.
REFERENCES
CDC (2012). Manual for the Surveillance of Vaccine–Preventable Diseases 5th Edition.
Dahiya S, Prasad G, Minakshi, Kovi RC (2005). Typing of bluetongue virus serotype 1 and 23 by RT–PCR. Indian J. Biotech. 4: 373–377.
Dalal S, Prasad G, Minakshi, Maan S (2008). VP7 gene based molecular characterization of two Indian isolates of bluetongue virus. Haryana Vet. 47: 41–45.
Dechamma HJ, Dighe V, Kumar CA, Singh RP, Jagadish M, Kumar S (2006). Identification of T–helper and linear B epitope in the hyper variable region of nucleocapsid protein of PPRV and its use in the development of specific antibodies to detect viral antigen. Vet. Microbiol.118:201 – 211.
http://dx.doi.org/10.1016/j.vetmic.2006.07.023
PMid:16962260
FAO (2000). Manual on meat inspection for developing countries.
Garg AK, Prasad G (1996). In vitro susceptibility of goat mononuclear cells to bluetongue virus. Indian J. Exp. Biol. 34: 818–820. PMid:8979494
Giridharan P, Hemadri D, Tosh C, Sanyal A, Bandyopadhyay SK (2005). Development and evaluation of a multiplex PCR for differentiation of foot–and–mouth disease virus strains native to India. J. Virol. Methods. 126(1–20): 1 – 11.
HariMohan, Minakshi, Kumar P, Prasad G (2012). Detection of Group B rotavirus in buffalo calves in Haryana state of Northern India. Indian J. Field Vet. 7: 71–74.
Jack PJ, Amos–Ritchie RN, Reverter A, Palacios G, Quan PL, Jabado O, Briese T, Lipkin WI, Boyle DB (2009). Microarray–based detection of viruses causing vesicular or vesicular–like lesions in livestock animals. Vet. Microbiol. 133 (1–2): 145–153.
http://dx.doi.org/10.1016/j.vetmic.2008.05.030
PMid:18621489
Jackwood MW, Hilt DA (1995). Production and immunogenicity of multiple antigenic peptide (MAP) constructs derived from the S1 glycoprotein of infectious bronchitis virus (IBV). Adv. Exp. Med. Biol. 380: 213 – 219.
http://dx.doi.org/10.1007/978-1-4615-1899-0_35
PMid:8830482
Joshi VG, Chindera K, Singh AK, Sahoo AP, Dighe VD, Thakuria D, Tiwari AK, Kumar S (2013a). Rapid label free visual assay for the detection and quantification of viral RNA using peptide nucleic acid (PNA) and gold nanoparticles (AuNPs). Anal.Chim.Acta. 17(795): 1–7.
http://dx.doi.org/10.1016/j.aca.2013.06.037
PMid:23998531
Joshi VG, Dighe VD, Thakuria D, Malik YPS, Kumar S (2013b). Multiple antigenic peptide (MAP): a synthetic peptide dendrimer for diagnostic, antiviral and vaccine strategies for emerging and re–emerging viral diseases. Indian J. Virol. 24(3): 312–320.
http://dx.doi.org/10.1007/s13337-013-0162-z
PMid:24426293
Killip MJ, Young DF, Precious BL, Goodbourn S, Randall RE (2012). Activation of the beta interferon promoter by paramyxoviruses in the absence of virus protein synthesis. J. General Virol. 93: 299 – 307.
http://dx.doi.org/10.1099/vir.0.037531-0
PMid:22049094 PMCid:PMC3352343
Kovi RC, Dahiya S, Prasad G, Minakshi (2007). Nucleotide sequence analysis of VP7 gene of Indian isolates of bluetongue virus vis–vis other serotypes from different parts of the world. DNA Seq, 17: 187–198.
http://dx.doi.org/10.1080/10425170600807264
Kumar P, Minakshi P, Ranjan K, Dalal R, Prasad G (2013). Evidence of reassortment between eastern and western topotype strains of bluetongue virus serotype 16 (BTV–16) from India. Adv. Anim. Vet. Sci. 1 (4S): 14 – 19.
Laemmli UK. (1970). Cleavage of structural proteins during the assembly of head of bacteriophage T4. Nature. 227: 680 – 685.
http://dx.doi.org/10.1038/227680a0
PMid:5432063
Mase M, Kenehira K (2012). Simple differentiation of avirulent and virulent strains of avian paramyxovirus serotype–1 (Newcastle disease virus) by PCR and restriction endonuclease analysis in Japan. J Vet Med Sci. 74(12): 1661–1664.
http://dx.doi.org/10.1292/jvms.12-0178
PMid:22814085
Minakshi P, Ranjan K, Kumar P, Prasad G (2013). A novel method of staining of RNA in polyacrylamide gel electrophoresis. Adv. Anim. Vet. Sci. 1 (4S): 20 – 23.
Minakshi P, Ranjan K., Pawan, Rupinder, Prasad G. (2011b). Segment 3 based genetic diversity among Indian isolates of bluetongue virus. Haryana vet. 50: 68 – 71.
Minakshi P, Singh R., Ranjan K., Kumar P, Joshi C.G, Reddy YKM, Prasad G (2012). Complete Genome Sequence of Bluetongue Virus Serotype 16 of Goat Origin from India. J. Virol. 86(15): 8337.
http://dx.doi.org/10.1128/JVI.01128-12
PMid:22787269 PMCid:PMC3421637
Minakshi P, Prasad G, Gupta A (2011a). Characterization of buffalo rotaviruses using DNA probes. Inter. J. Applied Engineering Res. 6: 687 – 690.
Minakshi, Prasad G, Malik Y, Pandey R (2005). G and P genotyping of bovine group A rotaviruses in fecal samples of diarrhoeic calves by DIG– labeled probes. Indian J. Biotech. 4: 93 – 99.
Minakshi, Prasad G, Sunita Verma, Swati Dahiya (2004). Detection of Group A avian rotaviruses from diarrhoeic poultry in India. Indian J. Microbiol. 44: 205 – 209.
Mishra I, Grover YP, Pandey R, Minakshi (2002). Comparison of detection limit of a non radioactive VP4 cDNA probe based dot blot hybridisation test and RNA–PAGE of cell culture grown bovine rotavirus. J. Immuno. Immunopatho.4: 33 – 39.
Naresh A, Roy P, Prasad G. (1996). Evaluation of recombinant bluetongue virus antigens using dot immunobinding assay. American J. Vet. Research. 57:1556 – 1558. PMid:8915428
Niture GS, Karpe AG, Minakshi P, Bhonsle AV, Ingale SS (2008). Genomic Diversity among Rotaviruses isolated from Diarrhoeic Buffalo calves. Vet. World. 2(7): 259 – 260.
OIE (2013). Manual of Diagnostic Tests and Vaccines for Terrestrial Animals.
Prasad G, Garg AK, Srivastava RN (1993). Electron microscopic observations on the morphogenesis and release of bluetongue virus from BHK 21 cells. Indian J. Virol. 9: 23 – 30.
Prasad G, Malik P, Malik P K, Minakshi (1998). Serological survey of bluetongue virus antibody in domestic and wild ruminants in and around Sariska tiger reserve, Rajasthan. Indian J. Virol. 14: 51– 53.
Prasad, G, Minakshi (1999). Comparative evaluation of sensitivity of RNA–polyacrylamide gel electrophoresis and dot immunobinding assay for detection of bluetongue virus in cell culture. Indian J. Exp. Biol. 37: 157 – 160.
PMid:10641136
Ramanathan A, Malik PK, Prasad G. (2007). Seroepizootiological survey for selected viral infections in captive Asiatic lions (Panthera leo persica) from western India. J. Zoo Wildl Med. 38(3): 400 – 408.
http://dx.doi.org/10.1638/2007-0006.1
PMid:17939348
Ranjan K, Prasad G, Kumar P, Minakshi P (2013). Vp5 gene based molecular characterization of bluetongue virus 9 from South India. Adv. Anim. Vet. Sci. 1 (4S): 30 – 36.
Ranjan K, Minakshi P, Mohan H, Rupinder, Vishwaradhya TM, Prasad G. (2012). Nucleotide sequence variation in vp5 gene of Indian isolate of bluetongue virus serotype 2. Harayana Vet. 51: 29 – 33.
Read SJ, Mitchell JL, Fink CG (2001). Light Cycler multiplex PCR for the laboratory diagnosis of common viral infections of the central nervous system. J. Clin. Microbiol. 39: 3056 – 3059.
http://dx.doi.org/10.1128/JCM.39.9.3056-3059.2001
PMid:11526128 PMCid:PMC88296
Roy P (2008). Bluetongue virus: dissection of the polymerase complex. J. Gen. Virol. 89: 1789 – 1804.
http://dx.doi.org/10.1099/vir.0.2008/002089-0
PMid:18632949 PMCid:PMC2735681
Satish S, Kumar A, Singh Y, Minakshi, Prasad G (2011). Identification and differentiation of raw meats of sheep, goat, cattle, pig and poultry by Multiplex PCR. Indian J. Anim. Sci. 81 (6): 588–591.
Savi J, Minakshi P, Prasad G (2009). Genotyping of field strains of canine parvovirus in Haryana using PCR and RFLP. Indian J. Anim. Sci.79: 71–73.
Smith LM, Sanders JZ, Kaiser RJ, Hughes P, Dodd C, Connell CR, Heiner C, Kent BH, Hood LE (1986). Fluorescence detection in automated DNA sequence analysis. Nature 321: 674 – 679.
http://dx.doi.org/10.1038/321674a0
PMid:3713851
Surendar S, Reddy YKM (2012). Development of multiplex RT–PCR for the determination of bluetongue virus serotypes. Tamilnadu J. Vet. Anim. Sci. 8 (2) :101–103.
Susmitha B, Sudheer D, Rao PP, Uma M, Prasad G, Minakshi P, Hegde NR, Reddy YN (2012). Evidence of bluetongue virus serotype 21 (BTV–21) divergence. Virus Genes. 44(3): 466–469.
http://dx.doi.org/10.1007/s11262-012-0724-y
PMid:22350945
Svensson L, Uhno I, Grandien M, Wadeli G (1986). Molecular epidemiology of rotavirus infections in Upsala. Sweden. 1981; disappearance of a predominant electropherotype. J. Med. Virol. 18: 101–111.
http://dx.doi.org/10.1002/jmv.1890180202
PMid:3005484
Tonnessen R, Kristoffersen A B, Jonassen CM, Hjortaas MJ, Hansen EF, Rimstad E, Hauge AG (2013). Molecular and epidemiological characterization of avian influenza viruses from gulls and dabbling ducks in Norway.Virol. J. 10: 112.
http://dx.doi.org/10.1186/1743-422X-10-112
PMid:23575317 PMCid:PMC3639200
Vishwaradhya TM, Minakshi P, Ranjan K, Supriya, Kumar P, Prasad G (2013). Sensitive detection of novel Indian isolate of BTV 21 using ns1 gene based real–time PCR assay. Vet. World 6(8): 554–557.
http://dx.doi.org/10.5455/vetworld.2013.554-557
Yu G, Zhang D, Guo F, Tan M, Jiang X, Jiang W (2013). Cryo–EM Structure of a Novel Calicivirus, Tulane Virus. PLoS ONE 8(3): e59817.
http://dx.doi.org/10.1371/journal.pone.0059817
PMid:23533651 PMCid:PMC3606144

