
Advances in Animal and Veterinary Sciences


Mini Review Article
Advances in Animal and Veterinary Sciences. 1 (4S): 1 – 4Special Issue–4 (Progress in Research on Viruses and Viral Diseases)
Diversity among Topotypes of Bluetongue Virus Serotype 9 as Revealed by Whole Genome Sequence Analysis
Sushila Maan1*, Arnab Ghosh1, Kanisth Batra1, Aman Kumar1, Narender Singh Maan2
- Department of Animal Biotechnology, College of Veterinary Sciences, LLR University of Veterinary and Animal Sciences, Hisar, 125 004, Haryana, India
- Department of Animal Nutrition, College of Veterinary Sciences, LLR University of Veterinary and Animal Sciences, Hisar, 125 004, Haryana, India
*Corresponding author:sushilamaan105@gmail.com
ARTICLE CITATION:
Maan S, Ghosh A, Batra K, Kumar A, Maan NS (2013). Diversity among topotypes of bluetongue virus serotype 9 as revealed by whole genome sequence analysis. Adv. Anim. Vet. Sci. 1 (4S): 1 – 4.
Received: 2013–09–29, Revised: 2013–11–05, Accepted: 2013–11–07
The electronic version of this article is the complete one and can be found online at
(
http://nexusacademicpublishers.com/table_contents_detail/4/128/html
)
which permits unrestricted use, distribution, and reproduction in any medium, provided the original work is properly cited
ABSTRACT
To understand the diversity and molecular epidemiology of Bluetongue virus serotype 9 (BTV–9), the complete genomes (19,177 base pairs) of several strains of BTV–9 originated from Europe, Greek islands, Mediterranean basin, Middle East, South Africa, Australia and Indian subcontinent were compared. These comparisons showed that all ten genome segments of a south African reference strain (RSArrrr/09) and its derivative vaccine strain (RSAvvvv/09) belong to western lineage, showing only 68% – 69% nucleotide (nt) identity with the eastern topotypic field strains including eastern strain from Australia (DP0837), Europe and Mediterranean region as well as reassortant strains from India. These detailed comparisons involving global strains showed that there is a very high degree of variation (up to 32%) between BTV–9 strains from eastern and western geographical regions. Full genome sequencing and availability of data are therefore highly recommended in order to quickly and accurately identify the emergence of viruses with novel genome segment constellation. These studies can also help to identify representative suitable ‘reference–strain’ of eastern topotype (BTV–9e), western topotype (BTV–9w), as well as ‘cross–topotype’ reassortant strains (BTV–9r) of BTV–9 which are circulating in the field.
Bluetongue (BT) is an arboviral disease of domestic and wild ruminants which is characterized by inflammation of the nasal and muzzle area, ulcers around the teeth and on the tongue, rapid weight loss, depression, diarrhea, and inflammation in the coronary band and laminar corium of the hoof resulting in lameness. The causative agent Bluetongue virus (BTV) is a double stranded RNA virus belonging to genus Orbivirus within family Reoviridae and is transmitted biologically between its vertebrate hosts (ruminants) by certain species of Culicoides biting midge (Diptera: Ceratopogonidae) (Mertens et al., 2005; Attoui et al., 2009).
Twenty six serotypes of BTV are now recognized globally (Hofmann et al., 2008; Maan et al., 2011a). BTV possesses a segmented genome comprising 10 segments of dsRNA which encode 11 proteins. Segmented viruses have the ability to ‘reassort’ genomic segments with related members of the same species (Shaw et al., 2013). The process of reassortment can cause fundamental shifts in the phenotypic characteristics of a virus. Reassortment event between pathogenic and non–pathogenic strains of a virus can lead to an increase in the pathogenicity of a previously avirulent strain (and vice versa) (Shelton et al., 2012). This phenomenon is of great importance with regard to the development of live attenuated vaccines for segmented viruses. The possibility exists for live attenuated vaccine and field viruses to reassert leading to viruses with a novel phenotype.
The size of the 10 genome segments of BTV–9 typically ranges from 822 to 3,944 bp. The lengths of these proteins (in amino acid residues (aa)) for BTV–9 are 1,302 (VP1), 955 (VP2), 901 (VP3), 644 (VP4), 526 (VP5), 330 (VP6), 552 (NS1), 354 (NS2), 229/ 216 (NS3/NS3A), and 77 (NS4), respectively (Grubman et al., 1983; Mertens et al., 1984; Belhouchet et al., 2011; Ratinier et al., 2011). These segments encode for seven structural and four nonstructural proteins observed during infection and replication. The virus core comprises the dsRNA genome segments associated with transcriptase complex of the virus which is made up of VP1 (RNA dependent RNA polymerase), VP4 (capping enzyme) and VP6 (the viral helicase), enclosed within consecutive layers of VP3 and VP7 (Grimes et al., 1998; Roy and Noad, 2006). The core is surrounded by an outer capsid layer comprising the variable proteins VP2 and VP5. NS1 is heavily expressed during infection and forms abundant cytoplasmic tubules which may be associated with cytopathogenicity (Owens et al., 2004). NS2 is an RNA–interacting phosphoprotein and is the major constituent of viral inclusion bodies (VIBs) where viral transcription and morphogenesis take place (Brookes et al., 1993). NS3/NS3A is a glycoprotein which is profoundly involved in virus egress and cell exit (Celma and Roy, 2009). NS4 is the most recently discovered BTV protein, which confers a replication advantage to cells pre–treated with interferon and has nucleolar localization (Belhouchet et al., 2011; Ratinier et al., 2011). Segments 2 and 5, which code for VP2 and VP5, determine the serotype of BTV (Maan et al., 2011b). Based on sequence differences, global isolates of BTV–9 can be grouped into eastern and western topotypes (Maan et al., 2009; Rao et al., 2012b).
More recently the global distribution of BTV has altered significantly with the introduction of novel serotypes into new areas, particularly in the northern hemisphere (Gibbs and Greiner, 1994; Saegerman et al., 2008; Tabachnick, 2010; Maclachlan, 2011; Maclachlan and Mayo, 2013). Thus, BT is regarded as a globally emerging disease (Purse et al., 2008; Maclachlan and Guthrie, 2010; Weaver and Reisen, 2010). Outbreaks of BT can be economically devastating to livestock production and the presence of BTV in a country can adversely impact the trade and movement of livestock (MacLachlan and Osburn, 2006; Velthuis et al., 2010; Rao et al., 2012). Predicting the course and geographic spread of an infectious disease is critical for its control. Control of BTV infection is difficult due to widely distributed Culicoides spp. midge vectors, the presence of vertebrate hosts and existence of a large number of serotypes of the virus. Prevention of the disease is characteristically dependent on vaccination of susceptible livestock or animal movement restrictions (Maclachlan and Mayo, 2013). Seg–2 was thus the original focus of sequence analyses for molecular epidemiology studies of BTV throughout the Mediterranean basin, northern Europe and elsewhere (Breard et al., 2003; Dahiya et al., 2004; Maan et al., 2004). However, whole genome sequencing studies have increasingly revealed evidence of reassortment between multiple strains of BTV in the field (Batten et al., 2008; Maan et al., 2008).
Whole genome sequencing (WGS) analysis studies that can thoroughly characterize the BTV type(s) circulating in a region and/or those involved in an outbreak, are necessary for the design and implementation of appropriate control strategies. Cross–reactive vaccines against multiple BTV strains/types would be of particular value in regions that are under the threat of incursions by multiple BTV serotypes. This article reviews the global distribution and overall genomic diversity as revealed by WGS analysis in the eastern and western topotypes of BTV–9.
BTV–9 was first isolated in Africa (Dungu et al., 2004), and later in Australia, South East Asia, and India (Pritchard et al., 2004; Prasad et al., 2009). Outbreaks caused by BTV–9 have been reported from the Mediterranean basin and Europe since 1998 (Saegerman et al., 2008). The first outbreaks of BT in Mediterranean basin since the 1980s were caused by BTV–9 and occurred on four Greek islands (Rhodes, Leros, Kos and Samos) during 1998. This was first report of BTV–9 in Europe, although there was earlier serological evidence for its presence in Turkey (Taylor and Mellor, 1994). Sequence analysis of Seg–2 demonstrated that this BTV–9 strain belongs to an eastern group of viruses, a finding that is consistent with its arrival in eastern Europe via Turkey, distinguishing it from the South African BTV–9 vaccine strains that belong to a western group (Maan et al., 2009). Using these sequence data, it has been possible to design primers that distinguish the European vaccine and field strains of BTV–9, by simply identifying their different topotypes (Mertens et al., 2007).
BTV–9, from the same eastern lineage, was identified as the cause of outbreaks in mainland Greece during 1999, south–eastern Bulgaria and European Turkey. During 2000, further outbreaks caused by BTV–9 were reported in north–western and central Greece, then in 2001 from Serbia, Montenegro, Kosovo, Macedonia, Bulgaria, Croatia, mainland Italy and Sicily. In 2002, BTV–9 was identified again in Bosnia, Bulgaria, Montenegro, Yugoslavia and Albania, and there was an unconfirmed report of BT in Kosovo (Calistri et al., 2004). BTV–9 was a major serotype which caused outbreaks from 2002 to 2006 in the state of Andhra Pradesh in India (Rao et al., 2012b).
Sequence comparisons of Seg–2 and Seg–6 showed that the isolates analyzed from the BTV–9 outbreaks in Europe are almost identical and can be grouped together as a single lineage (<12.9% nt sequence variation in Seg–2). All of these viruses belong to the same eastern geographic group as BTV–9 from Australia, India and Indonesia. BTV–9 had previously been reported in Anatolian Turkey, Syria, Jordan and Israel (Taylor and Mellor, 1994). Sequencing studies have shown >99% nt identity in Seg–2 between the BTV–9 field isolates from Italy 2001 and the Greek strains of BTV–9 from 1999, indicating that this serotype initially arrived in Italy from an easterly direction (Savini et al., 2004). Most of the European isolates of BTV–9 that have been characterized fully are distinct ‘eastern’ strains different from the South African reference and vaccine strains which belong to a distinct ‘western’ topotype of BTV–9 (Maan et al., 2009; Caporale et al., 2011). Three Indian isolate of BTV–9 from a southern state (Andhra Pradesh) of India (isolated in 2006 and 2007) have been fully characterized (Rao et al., 2012a; Rao et al., 2012b; Rao et al., 2013). These data have shown that these are reassortant strains, which have nine genome segments derived from eastern (e) lineage whereas Seg–5/NS1 gene from western (w) lineage strains. The co–circulation of a diverse array of BTV strains within the same population of animals and midges provides multiple opportunities for reassortment, resulting in the possibility of novel viruses with unknown or altered serological and/or pathogenic characteristics. Reassortment of an exotic virus having western Seg–5/NS1 gene with an endemic eastern strain of BTV–9 appears to favour the subsequent persistence of this gene from the exotic strain, which can be of key epidemiological significance in India.
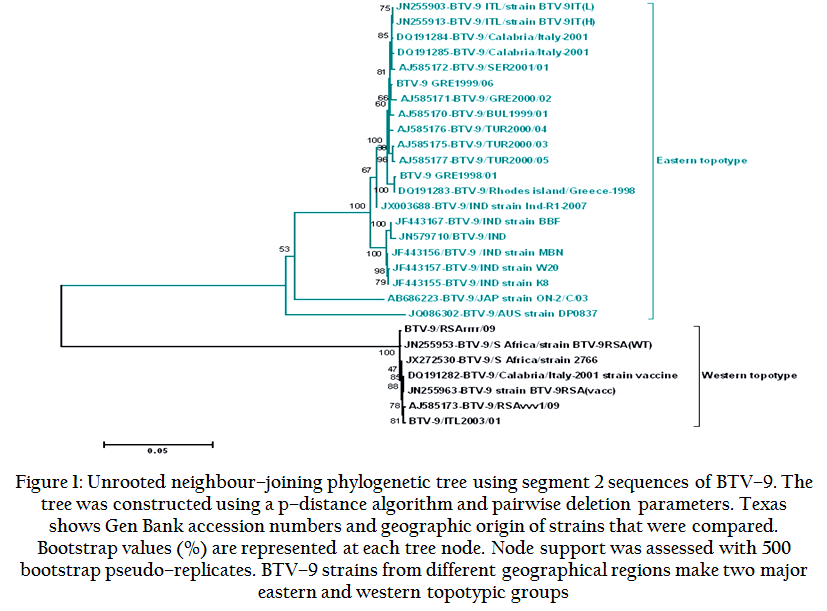
Figure 1: Unrooted neighbour–joining phylogenetic tree using segment 2 sequences of BTV–9. The tree was constructed using a p–distance algorithm and pairwise deletion parameters. Texas shows Gen Bank accession numbers and geographic origin of strains that were compared
Until now, seven eastern BTV–9 strains, from India, Australia and Italy along with two western strains, the South African reference and its derived vaccine strain, have been fully sequenced (Yang et al., 2011; Maan et al., 2012; Boyle et al., 2012). Previous analyses (Maan et al., 2012) show that the BTV–9 reference strain (RSArrrr/09), the BTV–9 vaccine strain (RSAvvvv/09) and a Sicilian BTV–9 (ITL2003/01) (Maan et al., 2009), which was recovered from an animal that died within a week after vaccination with the live BTV–9 vaccine, have >99% sequence identity in all ten genome segments, indicating that they are collectively derived from ‘a very recent common ancestor’. In contrast, BTV–9 from Australia (strain DP0837), represents a distinct virus lineage, although still grouping within the major eastern topotypic cluster (Figure 1). The Indian strains of BTV–9 are reassortants, showing high levels of nt identity in the majority of their genome segments to the European and Mediterranean strains but containing Seg–5 derived from a western topotype (with 98% nt identity South African BTV–3 reference virus) (Caporale et al., 2011; Rao et al., 2012b). Whole genome sequence comparisons also revealed that all ten genome segments of a south African reference strain (RSArrrr/09) and its derivative vaccine strain (RSAvvvv/09) shared only 68% – 69% nt identity in Seg–2 with the eastern topotypic field strains from Australia (DP0837) and reassortant strains from India. The Seg–6/VP5 of RSArrrr/09 and RSAvvvv/09 grouped within nucleotype C, different from eastern BTV–9 isolates including Indian reassortant strains which grouped within nucleotype B with nt/aa identity of 70 and 79%, respectively. These detailed comparisons involving global strains showed that there is a high degree of variation (up to 32%) between BTV–9 strains from eastern and western geographical regions.
CONCLUDING REMARKS
Results reviewed in this document contribute to the opinion that the epidemiological situation of BTV infections (including that of BTV–9) in Europe and Mediterranean basin illustrates the risk to the entire world of emerging diseases that were previously confined to specific geographic areas. Climate change and increased trade could be contributing factors for this changed scenario. From these detailed comparisons of BTV–9 strains it can be concluded that south African reference strain (RSArrrr/09), Australian strain (DP0837) and Indian strain (BEF) represented a suitable representative ‘reference–strain’ of BTV–9w, BTV–9e and BTV–9r for further serological, phylogenetic and molecular epidemiology studies (Mertens et al., 2013). Whole–genome sequencing and availability of these data facilitates quick and accurate identification of the emergence of viruses with novel genome segment constellation, which should be taken into account in BT control.
Author’s Contributions
SM, NSM: Substantial contribution in conception and design of experiments, acquisition and analysis of data, drafting the manuscript. AG, KB, AK: acquisition of data. All authors have read and approved the manuscript.
ACKNOWLEDGEMENTS
We are highly thankful to field veterinarians and all international colleagues who have provided virus isolates and data for these studies. This work was funded by Rastriya Krishi Vikas Yojna scheme no. 4011/C (g) ABT–4–0A.
COMPETING INTERESTS
The authors declared that they have no competing interests.
REFERENCES
Attoui H, Maan S, Anthony SJ and Mertens PPC (2009). Bluetongue virus, other Orbiviruses and other reoviruses: Their relationships and taxonomy. In: P.S. Mellor, M. Baylis and P.P.C. Mertens (eds). Bluetongue monograph, Elsevier/Academic Press, London, 1st ed: 23–46.
http://dx.doi.org/10.1016/B978-012369368-6.50007-1
Batten CA, Maan S, Shaw AE, Maan NS and Mertens PPC (2008). A European field strain of Bluetongue virus derived from two parental vaccine strains by genome segment reassortment. Virus Res. 137: 56–63.
http://dx.doi.org/10.1016/j.virusres.2008.05.016
PMid:18598726
Belhouchet M, Mohd Jaafar F, Firth AE, Grimes JM, Mertens PPC and Attoui H (2011). Detection of a Fourth Orbivirus Non–Structural Protein. PLoS ONE. 6(10): e25697.
http://dx.doi.org/10.1371/journal.pone.0025697
PMid:22022432 PMCid:PMC3192121
Boyle DB, Bulach DM, Amos–Ritchie R, Adams MM, Walker PJ and Weir R (2012). Genomic sequences of Australian Bluetongue virus prototype serotypes reveal global relationships and possible routes of entry into Australia. J. Virol. 86: 6724–6731
http://dx.doi.org/10.1128/JVI.00182-12
PMid:22514341 PMCid:PMC3393597
Breard E, Sailleau C, Coupier H, Mure–Ravaud K, Hammoumi S, Gicquel B, Hamblin C, Dubourget P and Zientara S (2003). Comparison of genome segments 2, 7 and 10 of Bluetongue viruses serotype 2 for differentiation between field isolates and the vaccine strain. Vet. Res. 34: 777–789.
http://dx.doi.org/10.1051/vetres:2003036
PMid:14746772
Brookes SM, Hyatt AD and Eaton BT (1993). Characterization of virus inclusion bodies in Bluetongue virus–infected cells. J. Gen. Virol. 74: 525–530.
http://dx.doi.org/10.1099/0022-1317-74-3-525
PMid:8383185
Calistri P, Giovannini A, Conte A, Nannini D, Santucci U, Patta C, Rolesu S and Caporale V (2004). Bluetongue in Italy: Part I. Vet. Ital. 40: 243–251.
PMid:20419672
Caporale M, Wash R, Pini A, Savini G, Franchi P, Golder M, Patterson–Kane J, Mertens P, Di Gialleonardo L, Armillotta G, Lelli R, Kellam P and Palmarini M (2011). Determinants of Bluetongue virus virulence in murine models of disease. J. Virol. 85: 11479–11489.
http://dx.doi.org/10.1128/JVI.05226-11
PMid:21865388 PMCid:PMC3194974
Celma CC and Roy P (2009). A viral nonstructural protein regulates Bluetongue virus trafficking and release. J. Virol. 83: 6806–6816.
http://dx.doi.org/10.1128/JVI.00263-09
PMid:19369335 PMCid:PMC2698550
Dahiya S, Prasad G and Kovi RC (2004). VP2 gene based phylogenetic relationship of Indian isolates of Bluetongue virus serotype 1 and other serotypes from different parts of the world. DNA Seq. 15: 351–361.
http://dx.doi.org/10.1080/10425170400012941
PMid:15621660
Dungu B, Potgieter C, Von Teichman B and Smit T (2004). Vaccination in the control of bluetongue in endemic regions: the South African experience. Dev. Biol. (Basel) 119: 463–472.
Gibbs EP and Greiner EC (1994). The epidemiology of bluetongue. Comp. Immunol. Microbiol. Infect. Dis. 17: 207–220.
http://dx.doi.org/10.1016/0147-9571(94)90044-2
Grimes JM, Burroughs JN, Gouet P, Diprose JM, Malby R, Zientara S, Mertens PP and Stuart DI (1998). The atomic structure of the Bluetongue virus core. Nature. 395: 470–478.
http://dx.doi.org/10.1038/26694
PMid:9774103
Grubman MJ, Appleton JA and Letchworth GJ (1983). Identification of Bluetongue virus type 17 genome segments coding for polypeptides associated with virus neutralization and intergroup reactivity. Virology. 131: 355–366.
http://dx.doi.org/10.1016/0042-6822(83)90503-2
Hofmann MA, Renzullo S, Mader M, Chaignat V, Worwa G and Thuer B (2008). Genetic characterization of toggenburg Orbivirus, a new Bluetongue virus, from goats, Switzerland. Emerg. Infect. Dis. 14: 1855–1861.
http://dx.doi.org/10.3201/eid1412.080818
PMid:19046507 PMCid:PMC2634640
Maan S, Maan NS, Nomikou K, Anthony SJ, Ross–smith N, Singh KP, Samuel AR, Shaw AE and Mertens PPC (2009). Molecular epidemiology studies of Bluetongue virus. In: P.S. Mellor, M. Baylis and P.P.C. Mertens (eds). Bluetongue monograph, Elsevier/Academic Press, London, Ist ed: 135–166.
Maan S, Maan NS, Nomikou K, Batten C, Antony F, Belaganahalli MN, Samy AM, Reda AA, Al–Rashid SA, El Batel M, Oura CA and Mertens PPC (2011a). Novel Bluetongue virus serotype from Kuwait. Emerg. Infect. Dis. 17: 886–889.
http://dx.doi.org/10.3201/eid1705.101742
PMid:21529403 PMCid:PMC3321788
Maan S, Maan NS, Nomikou K, Veronesi E, Bachanek–Bankowska K, Belaganahalli MN, Attoui H and Mertens PPC (2011b). Complete genome characterisation of a novel 26th Bluetongue virus serotype from kuwait. PLoS ONE. 6:e26147.
http://dx.doi.org/10.1371/journal.pone.0026147
PMid:22031822 PMCid:PMC3198726
Maan S, Maan NS, Ross–smith N, Batten CA, Shaw AE, Anthony SJ, Samuel AR, Darpel KE, Veronesi E, Oura CA, Singh KP, Nomikou K, Potgieter AC, Attoui H, van Rooij E, van Rijn P, De Clercq K, Vandenbussche F, Zientara S, Breard E, Sailleau C, Beer M, Hoffman B, Mellor PS and Mertens PPC (2008). Sequence analysis of Bluetongue virus serotype 8 from the Netherlands 2006 and comparison to other European strains. Virology. 377: 308–318.
http://dx.doi.org/10.1016/j.virol.2008.04.028
PMid:18570969
Maan S, Maan NS, Samuel AR, O'Hara R, Meyer AJ, Rao S and Mertens PPC (2004). Completion of the sequence analysis and comparisons of genome segment 2 (encoding outer capsid protein VP2) from representative isolates of the 24 Bluetongue virus serotypes. Vet. Ital. 40: 484–488.
PMid:20422574
Maan S, Maan NS, Singh KP, Belaganahalli MN, Guimera M, Pullinger G, Nomikou K and Mertens PPC (2012). Complete genome sequence analysis of a reference strain of Bluetongue virus serotype 16. J. Virol. 86: 10255–10256.
http://dx.doi.org/10.1128/JVI.00420-12
http://dx.doi.org/10.1128/JVI.00731-12
http://dx.doi.org/10.1128/JVI.00671-12
http://dx.doi.org/10.1128/JVI.01672-12
http://dx.doi.org/10.1128/JVI.00188-12
Maclachlan NJ (2011). Bluetongue: history, global epidemiology, and pathogenesis. Prev. Vet. Med. 102: 107–111.
http://dx.doi.org/10.1016/j.prevetmed.2011.04.005
PMid:21570141
Maclachlan NJ and Guthrie AJ (2010). Re–emergence of bluetongue, African horse sickness, and other Orbivirus diseases. Vet Res. 41: 35.
http://dx.doi.org/10.1051/vetres/2010007
PMid:20167199 PMCid:PMC2826768
Maclachlan NJ and Mayo CE (2013). Potential strategies for control of bluetongue, a globally emerging, Culicoides–transmitted viral disease of ruminant livestock and wildlife. Antiviral Res. 99: 79–90.
http://dx.doi.org/10.1016/j.antiviral.2013.04.021
PMid:23664958
MacLachlan NJ and Osburn BI (2006). Impact of Bluetongue virus infection on the international movement and trade of ruminants. J. Am. Vet. Med. Assoc. 228: 1346–1349.
http://dx.doi.org/10.2460/javma.228.9.1346
PMid:16649936
Mertens PPC, Brown F and Sangar DV (1984). Assignment of the genome segments of Bluetongue virus type 1 to the proteins which they encode. Virology. 135: 207–217.
http://dx.doi.org/10.1016/0042-6822(84)90131-4
Mertens PPC, Maan NS, Prasad G, Samuel AR, Shaw AE, Potgieter AC, Anthony SJ and Maan S (2007). Design of primers and use of RT–PCR assays for typing European Bluetongue virus isolates: differentiation of field and vaccine strains. J. Gen. Virol. 88: 2811–2823.
http://dx.doi.org/10.1099/vir.0.83023-0
PMid:17872535
Mertens PPC, Maan NS, Belaganahalli MN, Singh KP, Nomikou K and Maan S (2013). The full genome sequence of a western reference strain of Bluetongue virus serotype 16 from Nigeria. Genome Announc. 1(5):e00684–13.
http://dx.doi.org/10.1128/genomeA.00684-13
PMid:24051311 PMCid:PMC3778194
Mertens PPC, Maan S, Samuel A and Attoui H (2005). Orbiviruses, Reoviridae. In: C.M. Fauquet, M.A. Mayo, J. Maniloff, U. Desselberger and L.A. Ball, (eds). Virus Taxonomy. Eighth Report of the International Committee on Taxonomy of Viruses. Elsevier/Academic Press, London: 466–483.
Owens RJ, Limn C and Roy P (2004). Role of an arbovirus nonstructural protein in cellular pathogenesis and virus release. J. Virol. 78: 6649–6656.
http://dx.doi.org/10.1128/JVI.78.12.6649-6656.2004
PMid:15163755 PMCid:PMC416502
Prasad G, Sreenivasulu D, Singh KP, Mertens PPC and Maan S (2009). Bluetongue in the Indian subcontinent. In: P.S. Mellor, M. Baylis and P.P.C. Mertens (eds). Bluetongue monograph, Elsevier/Academic Press, London, Ist ed: 167–196.
Pritchard LI, Sendow I, Lunt R, Hassan SH, Kattenbelt J, Gould AR, Daniels PW and Eaton BT(2004). Genetic diversity of Bluetongue viruses in south east Asia. Virus Res. 101: 193–201.
http://dx.doi.org/10.1016/j.virusres.2004.01.004
PMid:15041187
Purse BV, Brown HE, Harrup L, Mertens PPC and Rogers DJ (2008). Invasion of bluetongue and other Orbivirus infections into Europe: the role of biological and climatic processes. Rev. Sci. Tech. 27: 427–442.
PMid:18819670
Rao PP, Hegde NR and Reddy YN (2012). Intercontinental movement of Bluetongue virus and potential consequences to trade. J. Virol. 86: 8341.
http://dx.doi.org/10.1128/JVI.00968-12
PMid:22787271 PMCid:PMC3421687
Rao PP, Reddy YN, Ganesh K, Nair SG, Niranjan V and Hegde NR (2013). Deep sequencing as a method of typing Bluetongue virus isolates. J. Virol. Methods. 193: 314–319.
http://dx.doi.org/10.1016/j.jviromet.2013.06.033
PMid:23831448
Rao PP, Reddy YN and Hegde NR (2012a). Complete genome sequence of Bluetongue virus serotype 9: implications for serotyping. J. Virol. 86: 8333.
http://dx.doi.org/10.1128/JVI.01101-12
PMid:22787266 PMCid:PMC3421634
Rao PP, Reddy YV, Meena K, Karunasree N, Susmitha B, Uma M, Prasad PU, Chaitanya P, Reddy YN and Hegde NR (2012b). Genetic characterization of Bluetongue virus serotype 9 isolates from India. Virus Genes. 44(2): 286–294.
http://dx.doi.org/10.1007/s11262-011-0707-4
PMid:22258368
Ratinier M, Caporale M, Golder M, Franzoni G, Allan K, Nunes SF, Armezzani A, Bayoumy A, Rixon F, Shaw A and Palmarini M (2011). Identification and characterization of a novel non–structural protein of Bluetongue virus. PLoS Pathog. 7:e1002477.
http://dx.doi.org/10.1371/journal.ppat.1002477
PMid:22241985 PMCid:PMC3248566
Roy P and Noad R (2006). Bluetongue virus assembly and morphogenesis. Curr. Top. Microbiol. Immunol. 309: 87–116.
http://dx.doi.org/10.1007/3-540-30773-7_4
http://dx.doi.org/10.1007/3-540-30773-7
PMid:16909898
Saegerman C, Berkvens D and Mellor PS (2008). Bluetongue epidemiology in the European Union. Emerg. Infect. Dis. 14: 539–544.
http://dx.doi.org/10.3201/eid1404.071441
PMid:18394269 PMCid:PMC2570923
Savini G, Potgieter AC, Monaco F, Mangana–Vougiouka O, Nomikou K, Yadin H and Caporale V (2004). VP2 gene sequence analysis of some isolates of Bluetongue virus recovered in the Mediterranean Basin during the 1998–2002 outbreak. Vet. Ital. 40: 473–478.
PMid:20422572
Shaw AE, Ratinier M, Nunes SF, Nomikou K, Caporale M, Golder M, Allan K, Hamers C, Hudelet P, Zientara S, Breard E, Mertens P and Palmarini M (2013). Reassortment between two serologically unrelated Bluetongue virus strains is flexible and can involve any genome segment. J. Virol. 87: 543–557.
http://dx.doi.org/10.1128/JVI.02266-12
PMid:23097432 PMCid:PMC3536370
Shelton H, Smith M, Hartgroves L, Stilwell P, Roberts K, Johnson B and Barclay W (2012). An influenza reassortant with polymerase of pH1N1 and NS gene of H3N2 influenza A virus is attenuated in vivo. J. Gen. Virol. 93: 998–1006.
http://dx.doi.org/10.1099/vir.0.039701-0
PMid:22323532 PMCid:PMC3541804
Tabachnick WJ (2010). Challenges in predicting climate and environmental effects on vector–borne disease episystems in a changing world. J. Exp. Biol. 213: 946–954.
http://dx.doi.org/10.1242/jeb.037564
PMid:20190119
Taylor WP and Mellor PS (1994). Distribution of Bluetongue virus in Turkey, 1978–81. Epidemiol. Infect. 112: 623–633.
http://dx.doi.org/10.1017/S0950268800051323
PMid:8005228 PMCid:PMC2271514
Velthuis AG, Saatkamp HW, Mourits MC, de Koeijer AA and Elbers AR (2010). Financial consequences of the Dutch bluetongue serotype 8 epidemics of 2006 and 2007. Prev. Vet. Med. 93: 294–304.
http://dx.doi.org/10.1016/j.prevetmed.2009.11.007
PMid:19962204
Weaver SC and Reisen WK (2010). Present and future arboviral threats. Antiviral Res. 85: 328–345.
http://dx.doi.org/10.1016/j.antiviral.2009.10.008
PMid:19857523 PMCid:PMC2815176
Yang T, Liu N, Xu Q, Sun E, Qin Y, Zhao J and Wu D (2011). Complete genomic sequence of Bluetongue virus serotype 16 from China. J. Virol. 85(24): 13472.
http://dx.doi.org/10.1128/JVI.06402-11
PMid:22106384 PMCid:PMC3233117

