{kind=link}
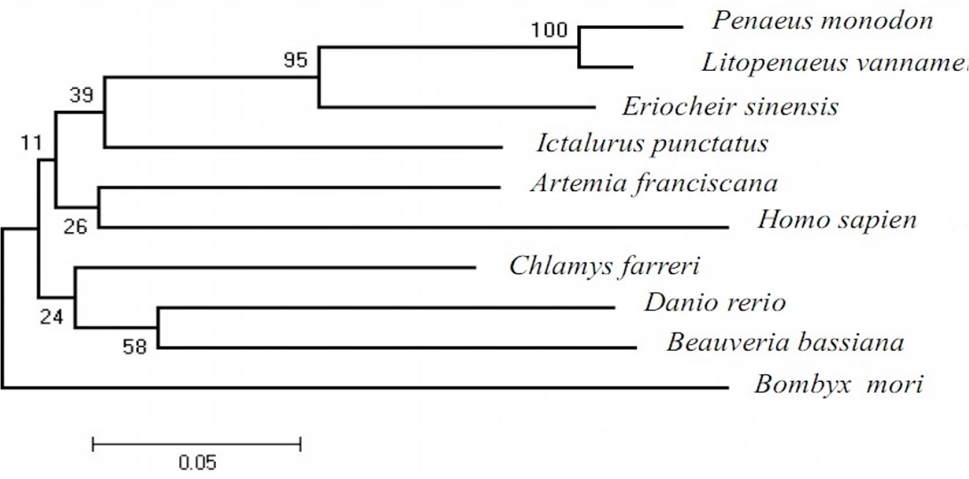
Fig 3
A phylogenetic tree constructed with the neighbor- joining method. The tree is based on an alignment corresponding to full length amino acid sequences, using ClustalX and megAlign. The numbers shown at the branches denote bootstrap majority consensus values of 1000 replicates. The GenBank accession numbers are same as given in Figure 2.